
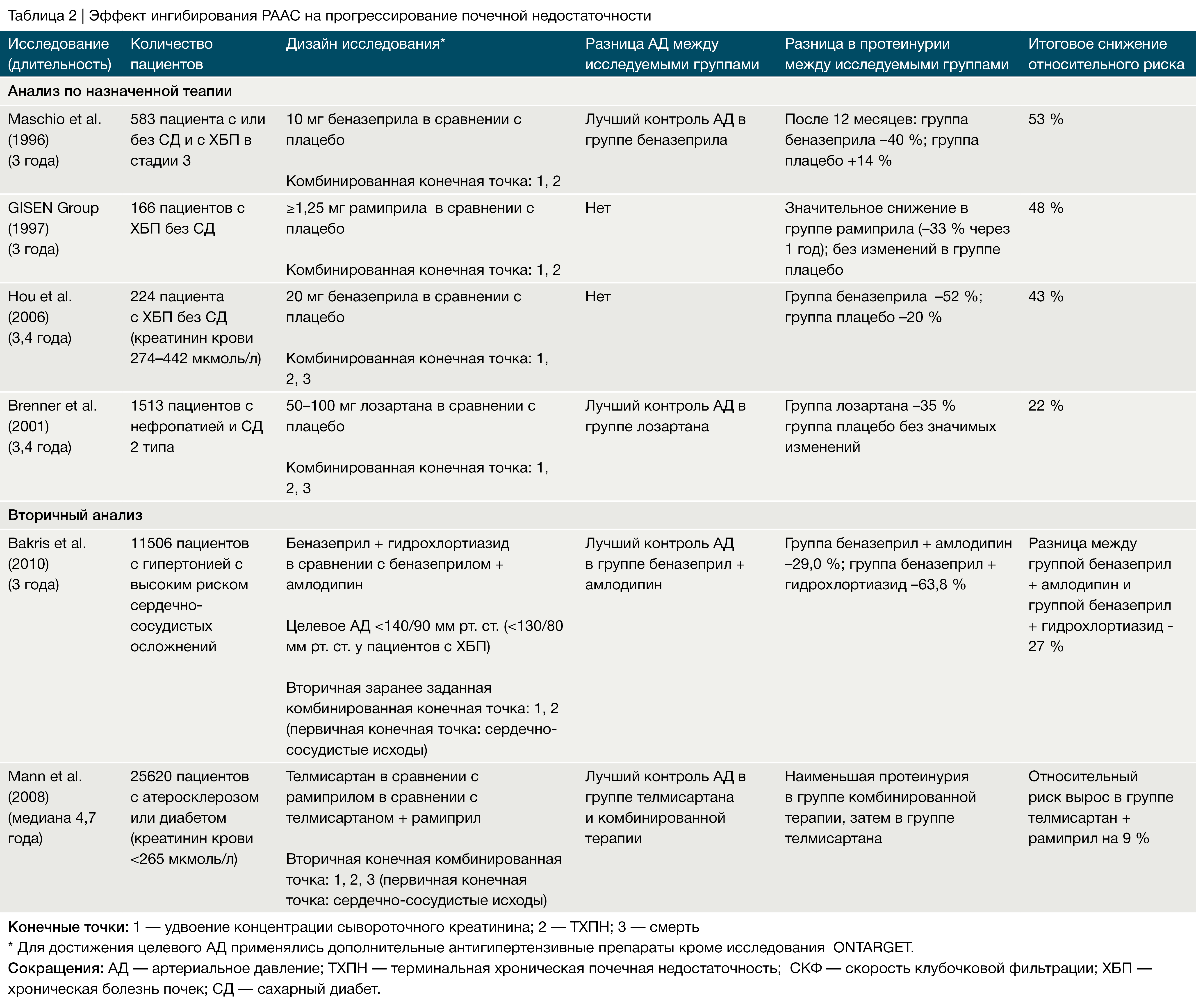
Контроль артериального давления в ведении хронической болезни почек

Абстракт | Прогрессирование хронической болезни почек (ХБП) при достижении критического уровня функции почек уже мало зависит от лежащего в основе заболевания. Гипертония является независимым фактором риска как у детей, так и у взрослых. В настоящее время внимание исследователей сосредоточено на блокаде ренин‑ангиотензин‑альдостероновой системы (РААС) и уже накоплено достаточно доказательств эффективности медикаментозной нефропротекции.
Ингибиторы ангиотензинпревращающего фермента (иАПФ) и блокаторы рецепторов ангиотензина (БРА, сартаны) обеспечивают эффективный контроль не только артериального давления, но и протеинурии, что связано с лучшей нефропротекцией по сравнению с другими классами антигипертензивных препаратов. Кроме того, появляются данные о дополнительном нефропротекторном преимуществе строгого контроля артериального давления (АД) с целевым АД в пределах субнормального у молодых пациентов и пациентов с протеинурией. В этом обзоре авторы опишут роль гипертонии в патогенезе ХБП и обсудят основные принципы терапии, а также роль антигипертензивных препаратов в предотвращении прогрессирования почечной недостаточности.
Введение
Прогрессирование почечной недостаточности до терминальной стадии — распространенное явление у больных ХБП. При достижении определенного критического ухудшения почечной функции происходит дальнейшее ее ускоренное снижение независимо от основного заболевания почек. Так как терминальная хроническая почечная недостаточность (ТХПН, 5 стадия ХБП) сопровождается высокой заболеваемостью и летальностью, то эффективная терапия, направленная на замедление прогрессирования ХБП и отсроченное начало заместительной почечной терапии, повлияет на качество и продолжительность жизни пациентов. Следовательно, изучение и равзвитие нефропротективных стратегий является актуальным направлением исследований ХБП у детей и взрослых.
Экспериментальные и клинические исследования предоставили большой объем доказательств того, что гипертония и протеинурия являются не только маркерами, но и ключевыми патофизиологическими механизмами прогрессирования ХБП [1–3]. Ренин-ангиотензин-альдостероновая система, по‑видимому, играет ключевую роль в этом процессе. В меньшей степени прогрессированию ХБП способствуют дислипидемия, воспаление и оксидативный стресс, анемия и нарушения минерального обмена. Более того, основные факторы риска сердечно-сосудистых заболеваний (ожирение, диабет и курение) и генетика являются важными дополнительными детерминантами, влияющими на функцию почек. В этом обзоре описаны основные принципы терапии, направленной на сохранение функции почек. Авторы уделяют особое внимание роли гипертонии в прогрессировании ХБП, а также антигипертензивной терапии в предотвращении прогрессирования ХБП у детей и взрослых.
Классическое течение ХБП
ХБП чаще страдают взрослые, в том числе более 10 % пожилого населения. Наиболее распространенными причинами ХБП у взрослых являются диабетические и гипертензивные гломерулопатии, за ними следуют IgA‑нефропатия, фокальный сегментарный гломерулосклероз, поликистоз почек и рефлюксная нефропатия (гидронефроз). Скорость прогрессирования ХБП вариабельна и зависит от ряда факторов, в том числе от основного заболевания, наличия или отсутствия сопутствующих заболеваний, терапии, социально-экономического статуса, генетической предрасположенности, этнической принадлежности и других факторов.
ХБП гораздо реже встречается у детей (менее 100 случаев на миллион). ХБП у детей обычно вызвана врожденными аномалиями почек и мочевыводящих путей или наследственными гломерулопатиями. Изучению течения ХБП в педиатрии посвящено не так много исследований. В популяционном исследовании детей с врожденными аномалиями почек и мочевыводящих путей (ItalKid Study) [5], риск развития ТХПН к 20 годам составил 68 %. На начальных этапах при врожденных аномалиях почек происходит гипертрофия нефронов, при которой скорость клубочковой фильтрации (СКФ) может значительно увеличиться. Этот период может длиться несколько лет и обычно характеризуется достаточной функцией почек, но в конечном итоге СКФ необратимо падает до уровня ТХПН [6]. У многих пациентов происходит значительное прогрессирование ХБП во время полового созревания [6].
Патофизиология прогрессирования ХБП
Современная концепция патогенеза ХБП, сформулированная Бреннером [7], гласит: любая критическая потеря функциональной массы почек, независимо от характера первоначального повреждения, приводит к гломерулярной гиперфильтрации и, соответственно, к повышению СКФ оставшихся нефронов. Согласно этой гипотезе, которая была исследована на модели крысиной почки и подтверждена для некоторых болезней и состояний у человека (таких как врожденная или приобретенная одиночная почка) [8], оставшиеся нефроны теряют способность к ауторегуляции гломерулярного давления, что приводит к передаче системной гипертензии напрямую в клубочек и вызывает гломерулярную и тубулярную гипертрофию. Повреждение клеток эндотелия и подоцитов, вызванное какими-либо специфическими или неспецифическими заболеваниями, например, васкулотоксическим и воспалительным влиянием мочевины, часто является звеном патогенеза в прогрессировании повреждения клубочков, индуцируя местное воспаление и фиброз [9–11]. Кроме того, протеинурия, индуцированная увеличением гломерулярного давления, считается патофизиологическим связующим звеном между гломерулярным, интерстициальным и тубулярным повреждениями [12, 13]. Степень протеинурии при гломерулярных заболеваниях коррелирует со скоростью прогрессирования почечной недостаточности [14]. Реабсорбция белков тубулоэпителиальными клетками может вызвать прямое нарушение работы лизосомальных систем, оксидативный стресс, локальное повышение экспрессии факторов роста [15, 16] и высвобождение хемотаксических факторов, которые способствуют тубулоинтерстициальному воспалению и фиброзу путем рекрутирования и активации макрофагов [17–22].
Макрофаги, инфильтрирующие почечную паренхиму, в свою очередь, сами начинают продуцировать цитокины и факторы роста. И в клубочках, и в канальцах хроническое воспаление приводит к усилению синтеза и снижению деградации внеклеточного матрикса с чрезмерным накоплением тубулоинтерстициального коллагена. Последующие гломерулярный склероз, тубулоинтерстициальный фиброз и атрофия почечных канальцев вызывают дальнейшую потерю функционирующей почечной массы, тем самым замыкая порочный круг заболевания, увеличивая внутриклубочковое давление и гипертрофию оставшихся клубочков.
Ангиотензин II — основной эффектор РААС, непосредственно участвует в большинстве описанных выше процессов. Ангиотензин II продуцируется как системно, так и локально в почках и имеет множество эндокринных, аутокринных и паракринных эффектов. Его внутрипочечная концентрация на три порядка выше, чем в системном кровотоке, и поддерживается аутокринными и паракринными механизмами гормонального синтеза тубулярными, юкстагломерулярными и клубочковыми клетками [23–25]. Большинство внутрипочечных эффектов ангиотензина II опосредуются через рецепторы ангиотензина II первого типа [26]. Ангиотензин II является мощным вазоконстриктором, который увеличивает внутриклубочковое давление преимущественно путем увеличения тонуса эфферентных артериол. Он также увеличивает концентрацию кальция в подоцитах [27], вызывая изменения цитоскелета и функции подоцитов, тем самым индуцируя ультрафильтрацию белка даже в отсутствие структурного повреждения клубочков [28, 29]. Кроме того, ангиотензин II стимулирует пролиферацию клеток гладкой мускулатуры и усиливает гломерулярную и канальцевую экспрессию различных факторов роста, цитокинов и хемокинов, стимулируя оксидативный стресс, который сам по себе повышает активность цитокинов, молекул адгезии и хемоаттрактантов [30–31], тем самым вновь замыкая патологический механизм хронического воспаления. Наконец, внутрипочечный ангиотензин II стимулирует афферентную импульсацию, предположительно активирующую ответственные за регуляцию симпатического тонуса структуры центральной нервной системы. Таким образом, ангиотензин II участвует в патофизиологической симпатической гиперактивации, что характерно для ХБП, и является еще одним важным механизмом прогрессирования почечной недостаточности и заболеваний сердечно-сосудистой системы [32, 33].
Артериальная гипертензия
Артериальная гипертензия — крайне важный независимый фактор риска прогрессирования ХБП [1, 3, 4]. Тяжесть гипертонии не только отражает активность болезни почек, но и, как указано выше, также считается причиной прогрессирования ХБП. В одном из мета‑анализов [34] девяти крупных клинических исследований у пациентов с диабетической и недиабетической почечной недостаточностью была отмечена сильная линейная корреляция между достигнутым уровнем артериального давления и степенью прогрессирования ХБП: более низкое давление замедляло прогрессию ХБП. В исследовании RENAAL повышение систолического АД на каждые 10 мм рт. ст. от исходного уровня увеличивало риск развития ТХПН или смерти на 6,7 % [35]. Интересно, что пациенты, имеющие самые высокие цифры АД, имели, соответственно, самый высокий риск прогрессирования ХБП, но также и наибольшее уменьшение риска при снижении уровня АД ниже 140 мм рт. ст.
Независимо от абсолютного уровня АД, ослабление физиологического ночного снижения АД («диппинга») может играть не последнюю роль в прогрессировании ХБП. Это состояние («нон‑диппинг») является фактором риска сердечно‑сосудистых заболеваний [36] и характерным проявлением ренопаренхимальной гипертензии; было высказано предположение, что оно отражает состояние симпатической гиперактивации, которая характерна для ХБП [32, 33, 37]. С другой стороны, восстановление суточной ритмичности динамики АД с помощью ограничения потребления натрия или утреннего приема диуретиков у пациентов с соль-чувствительной гипертензией или ХБП говорит о соль-зависимом механизме развития артериальной гипертензии [38]. В нескольких работах была предложена связь нон-диппинга с более быстрой потерей функции почек у пациентов с ХБП [39, 40]. Однако исключить искажение под влиянием конкурирующих факторов риска в обсервационных исследованиях трудно; в 2009 году в анализе пациентов пожилого возраста с или без ХБП связи циркадных изменений уровня АД со смертностью или ТХПН не наблюдалось после учета возраста, гипертонии, СКФ и протеинурии [41]. Тезис, что успешное восстановление циркадного ритма АД благодаря фармакологическому вмешательству имеет долгосрочную клиническую пользу, с точки зрения сердечно-сосудистой или нефропротективной терапии все еще ожидает своего подтверждения в будущих клинических испытаниях.
Протеинурия — дополнительный фактор риска
Поскольку гипертония, гломерулярная гиперфильтрация, повреждение почек и протеинурия тесно взаимосвязаны, необходимо также учитывать влияние протеинурии в контексте прогрессирования ХБП. Популяционные исследования также определили протеинурию как независимый фактор развития ТХПН и повышения общей смертности [42–44]. У пациентов как с диабетической, так и с недиабетической нефропатией экскреция белка с мочой дает четкое представление о дальнейшей динамике функции почек [14, 45, 46]. В исследовании REIN у взрослых пациентов с недиабетической ХБП наличие протеинурии в начале исследования было единственным параметром, ассоциированным со снижением СКФ вплоть до ТХПН [47]. Несмотря на разнообразный спектр заболеваний, лежащих в основе ХБП у детей, протеинурия также является главным предиктором снижения СКФ при нефропатиях в педиатрической практике [48, 49].
Раннее снижение протеинурии посредством консервативной терапии отчетливо замедляет скорость снижения СКФ, что было доказано в пролонгированных исследованиях [14, 50–52]. В исследовании MDRD снижение суточной протеинурии на каждый грамм в течение 4 месяцев посредством антигипертензивной терапии и/или низкопротеиновой диеты давало замедление снижения СКФ на 1 мл/мин в год [14].
В исследовании REIN ежегодное снижение СКФ замедлилось на 2 мл/мин на каждый сниженный грамм протеинурии в ходе 3 месяцев терапии ингибиторами АПФ [53]. В педиатрическом исследовании ESCAPE сохранение функции почек было ассоциировано с остаточной протеинурией на ингибиторах АПФ (Рис. 1) [54]. Максимальное снижение риска прогрессирования ХБП предположительно связано с уменьшением суточной протеинурии до < 300 мг/м2 в день [55, 56]. Следует, однако, подчеркнуть, что доказательства пользы агрессивной фармакотерапии протеинурии в настоящее время основаны на post hoc-анализе исследований, в которых снижение протеинурии не было основной терапевтической целью. Поэтому требуется проведение клинических исследований, оценивающих пользу интенсивного контроля протеинурии.
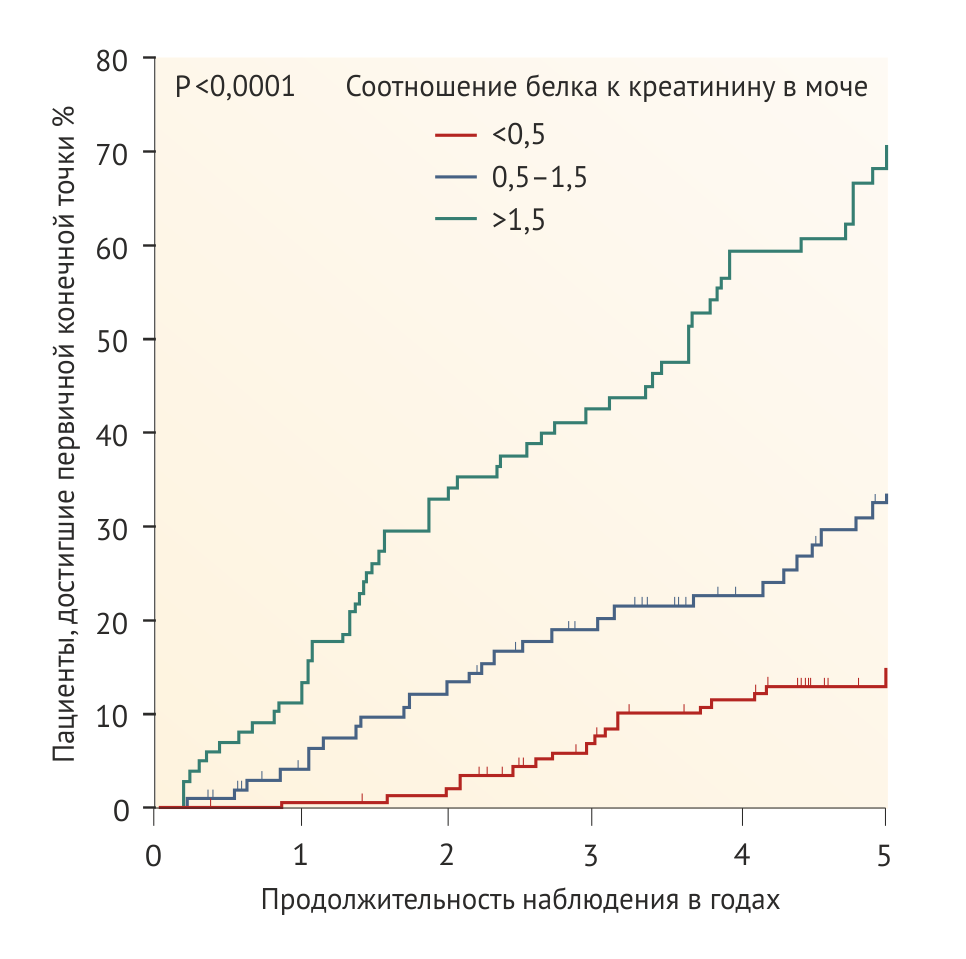
Контроль АД и ХБП: нефропротекция
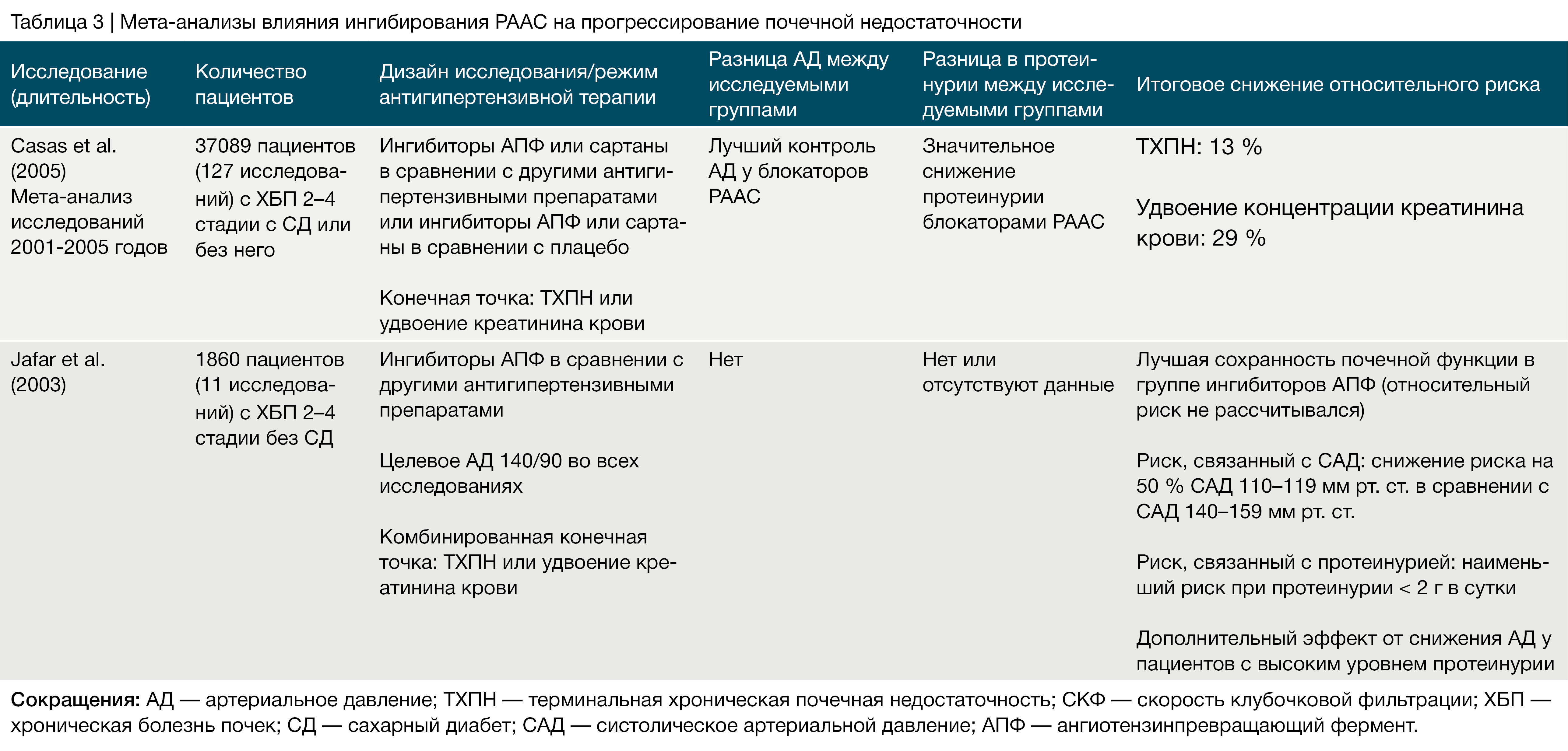
В последние несколько лет появились фармакологические стратегии нефропротекции, основанные на глубоком понимании прогрессирования почечной болезни. Эффективный и устойчивый контроль АД и минимизация протеинурии используются в качестве промежуточных конечных точек для прогнозирования долговременного эффекта. Многочисленные исследования подтвердили концепцию, согласно которой антигипертензивная терапия замедляет скорость прогрессирования ХБП [14, 34, 57]. Мета‑анализ результатов исследований по контролю АД у пациентов с ХБП показал взаимосвязь между достигнутым уровнем АД и скоростью прогрессирования почечной недостаточности [34], которая, по‑видимому, сохраняется в нормальном диапазоне артериального давления [14, 57].
Такая сильная связь между контролем артериального давления и сохранностью функции почек у пациентов с ХБП привела к снижению рекомендованных показателей артериального давления. Последние американские [58] и европейские [59] рекомендации по гипертонии определили уровень АД 120/80 мм рт. ст. в качестве верхнего предела «оптимального» диапазона АД для пациентов с ХБП и предположили, что уровень артериального давления > 130/80 мм рт. ст. следует снизить с помощью консервативной терапии. Эти значения соответствуют 50–75 процентилям в генеральной совокупности взрослых людей.
Результаты рандомизированного контролируемого исследования ESCAPE 2009 года показали, что у детей с ХБП в стадии 2–4 антигипертензивная терапия, направленная на круглосуточное поддержание давления в диапазоне ниже нормального (то есть ниже 50-го возрастного процентиля) улучшает выживаемость у таких пациентов по сравнению с теми, у кого целевой уровень АД был в промежутке между 50-м и 95-м процентилями [54]. Все пациенты в этом исследовании получали одинаковую фиксированную дозу ингибитора АПФ, а дополнительное снижение АД в исследуемой группе производилось посредством добавления к их терапии других лекарств, таких как блокаторы медленных кальциевых каналов (БМКК), диуретики, α- и β-адреноблокаторы. Несмотря на относительно небольшое снижение уровня АД (примерно на 3–4 мм рт. ст.), почечная функция заметно улучшилась благодаря более интенсивной антигипертензивной терапии.
Установка низкого нормального целевого АД в рандомизированной группе привела к снижению на 35 % риска сокращения СКФ до 50 % или прогрессирования в ТХПН в течение 5 лет (Рис. 2, Табл. 1). Хотя строгий контроль артериального давления казался наиболее эффективным именно у пациентов до достижения пубертатного возраста с артериальной гипертонией и/или протеинурией, у которых прогрессировала ХБП, преимущества этого подхода сохранялись и в других группах пациентов. Усиленный контроль АД достоверно улучшает прогноз почечной функции у детей с гломерулопатиями или дисплазией почек, в то же время это утверждение не является верным в отношении других врожденных или наследственных нефропатий [54].
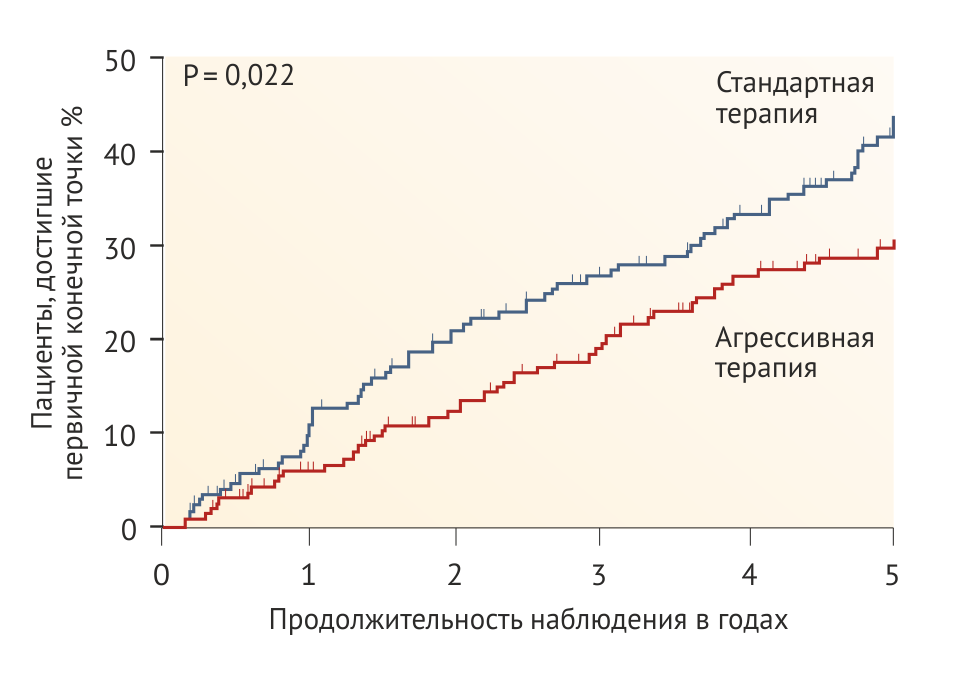
Преимущество строгого контроля АД у детей продемонстрировало исследование ESCAPE [54], что соотносится с результатами исследования MDRD у взрослых пациентов с ХБП и протеинурией [60], но отличается от результатов других исследований у взрослых пациентов (Табл. 1–3). В исследовании MDRD пациенты с протеинурией, попавшие в группу с низким целевым значением АД (среднее АД < 92 мм рт. ст.), демонстрировали лучшую сохранность почечной функции после 4 лет наблюдения, тогда как у пациентов без протеинурии, у которых целевое АД также было ниже среднего, данная тактика не показала каких-либо преимуществ [60].
Долгосрочное наблюдение более 10 лет выявило стойкое снижение риска прогрессирования почечной недостаточности в группе низкого АД, в то же время влияние протеинурии потеряло статистическую значимость [57]. Исследование MDRD могло быть необъективно из-за преимущественного применения иАПФ, тогда как в исследовании ESCAPE использование анти-РААС терапии было стандартизировано и идентично для обеих исследуемых групп [54].
В расширенном исследовании REIN-2 добавление 5–10 мг фелодипина к 2,5–5 мг рамиприла в день взрослым пациентам с ХБП, протеинурией и без диабета в группе с целевым уровнем АД < 130/80 мм рт. ст. по сравнению с контрольной группой, у которой целевое значение диастолического АД было < 90 мм рт. ст., не привело к улучшению сохранности функции почек, хотя чувствительность исследования была ограничена кратковременной продолжительностью наблюдения (медиана 19 месяцев) [61]. В исследовании AASK участвовали более 1000 афроамериканцев с гипертоническим нефросклерозом, которые были случайным образом разделены на группы, получающие рамиприл, метопролол или амлодипин, а также на группы с целевым средним АД 102–107 мм рт. ст. или < 90 мм рт. ст. Влияние на скорость снижения СКФ не зависело от принимаемого препарата [62]. Однако на этапе наблюдения в группе пациентов, принимающих рамиприл, строгий контроль АД оказывал нефропротективный эффект в подгруппе пациентов с исходной протеинурией более 0,22 мг на мг креатинина [63]. Это подтверждает концепцию, согласно которой жесткий контроль артериального давления может улучшить функцию почек у афроамериканцев‑гипертоников с ХБП и протеинурией. В исследовании ABCD у пациентов с артериальной гипертензией низкое целевое АД не влияло на сохранность почечной функции, тогда как пациенты с нормотензией, по‑видимому, имели некоторое преимущество от снижения уровня АД до низких нормальных значений [64].
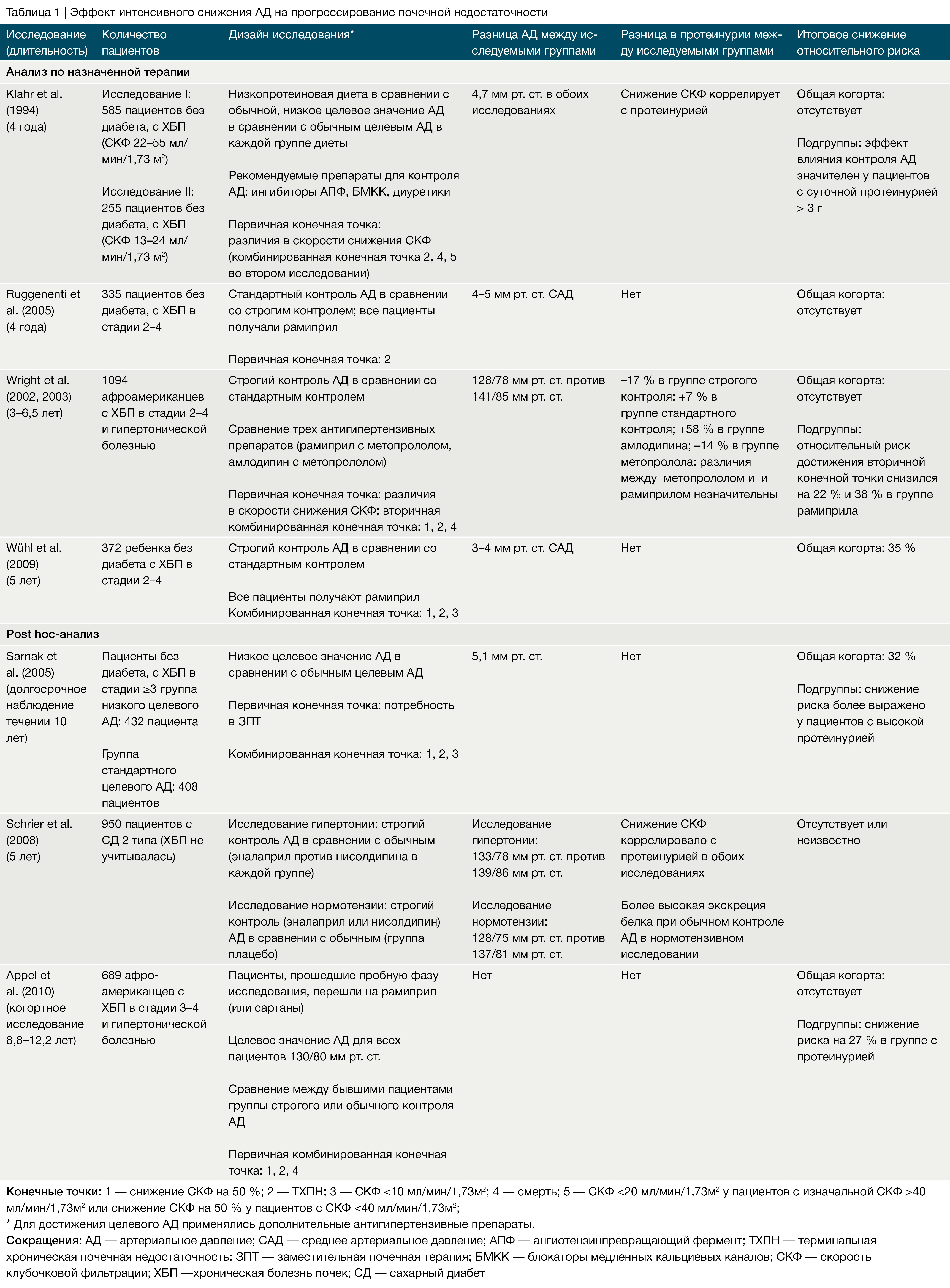
Некоторое расхождение результатов в различных исследованиях исходов ХБП может быть объяснено методологическими и демографическими различиями. Возраст пациентов, этническая принадлежность, протоколы фармакологического лечения, продолжительность наблюдения и количество вышедших из исследования пациентов значительно варьировались между упомянутыми выше исследованиями (Табл. 1–3). Кроме того, применение амбулаторного контроля АД, возможно, могло обеспечить более чувствительный мониторинг достигнутого уровня АД в исследовании ESCAPE у детей, нежели у взрослых пациентов в других исследованиях, в которых изучались показатели АД в бытовых условиях [65].
Низкие целевые значения АД хорошо переносятся большинством пациентов. Что касается сердечно-сосудистых исходов, феномен «J-кривой» (небольшое увеличение сердечно-сосудистых событий у пациентов с очень низким уровнем артериального давления), скорее всего, относится к пожилым пациентам с тяжелым атеросклерозом. Это явление может объяснить результаты исследования ONTARGET у пожилых пациентов с диабетом и с высоким уровнем риска сердечно-сосудистых осложнений. В этом исследовании строгий контроль АД был связан с высоким риском достижения (вторичной) составной «почечной» конечной точки, определяемой как увеличение вдвое уровня креатинина в сыворотке крови, потребность в диализе или смерть. Тем не менее, именно этой конечной точки достигли 86 % пациентов из тех 12 %, которые вышли из исследования по достижении составной конечной точки.
Влияние на протеинурию
Исследования MDRD [14], ABCD [67] и AASK [51] продемонстрировали, что эффективный контроль АД также снижает протеинурию. Низкое целевое значение АД (< 125/75 мм рт. ст. у взрослых) снижало абсолютные цифры протеинурии на 50 % [14] и предотвращало двух-трехкратное нарастание протеинурии у пациентов со стандартным целевым АД (140/90 мм рт. ст.) [67]. Аналогичным образом в исследовании ESCAPE эффективное снижение АД у детей с помощью иАПФ было связано с уменьшением протеинурии на 50 % в течение 6 месяцев лечения [54]. Ранний антипротеинурический ответ является хорошим предиктором долгосрочной сохранности почечной функции в соответствии с предыдущими результатами исследований у взрослых.
Фармакологические препараты в терапии гипертонии
Антагонисты РААС
Большинство классов антигипертензивных препаратов более или менее сходны по влиянию на уровень АД, чего нельзя сказать в отношении их влияния на протеинурию и прогрессирование ХБП [51, 55, 70, 71]. Ингибиторы АПФ и сартаны являются препаратами выбора у детей и взрослых с ХБП. В терапии эссенциальной гипертензии иАПФ обеспечивают наилучшее качество жизни по сравнению с другими классами препаратов [72] благодаря их превосходному профилю безопасности, сравнимому с плацебо.
Несколько рандомизированных исследований показали, что у пациентов с диабетом или без применение иАПФ или сартанов обеспечивает снижение протеинурии на 30–40 % по сравнению с плацебо или другими антигипертензивными препаратами [55, 71, 73, 74]. В пролонгированных исследованиях этот эффект был связан с более медленным снижением СКФ [50, 55, 75–83]. А вот в отношении долгосрочной нефропротекции роль препаратов, влияющих на РААС, остается спорной [84]. Несмотря на то, что риск прогрессирования ХБП уменьшается на 30–40 %, эффективность этих препаратов зависит в первую очередь от уровня изначальной протеинурии. У взрослых пациентов необходимая эффективность достигается при протеинурии более 500 мг в день [55].
У некоторых пациентов во время лечения развивается вторичная резистентность к гипопротеинурическому эффекту иАПФ (Рис. 3) [54, 85–87]. Вероятно, это связано с феноменом прорыва альдостерона, наблюдаемым почти у 40 % взрослых пациентов, получающих длительную терапию иАПФ [88]. Предположительно, в основе этого явления лежит активация ферментов, ускользающих от влияния иАПФ, например, химазы. С другой стороны, внутрипочечные вазоактивные медиаторы могут компенсировать недостаток влияния ангиотензина, а отсроченный рост протеинурии отражает естественное течении патологии, лежащей в основе почечной недостаточности. Независимо от того, какой механизм был задействован, уровень остаточной протеинурии после лечения обратно пропорционален сохранению функции почек [54] (Рис. 1) и скорости снижения СКФ [50].
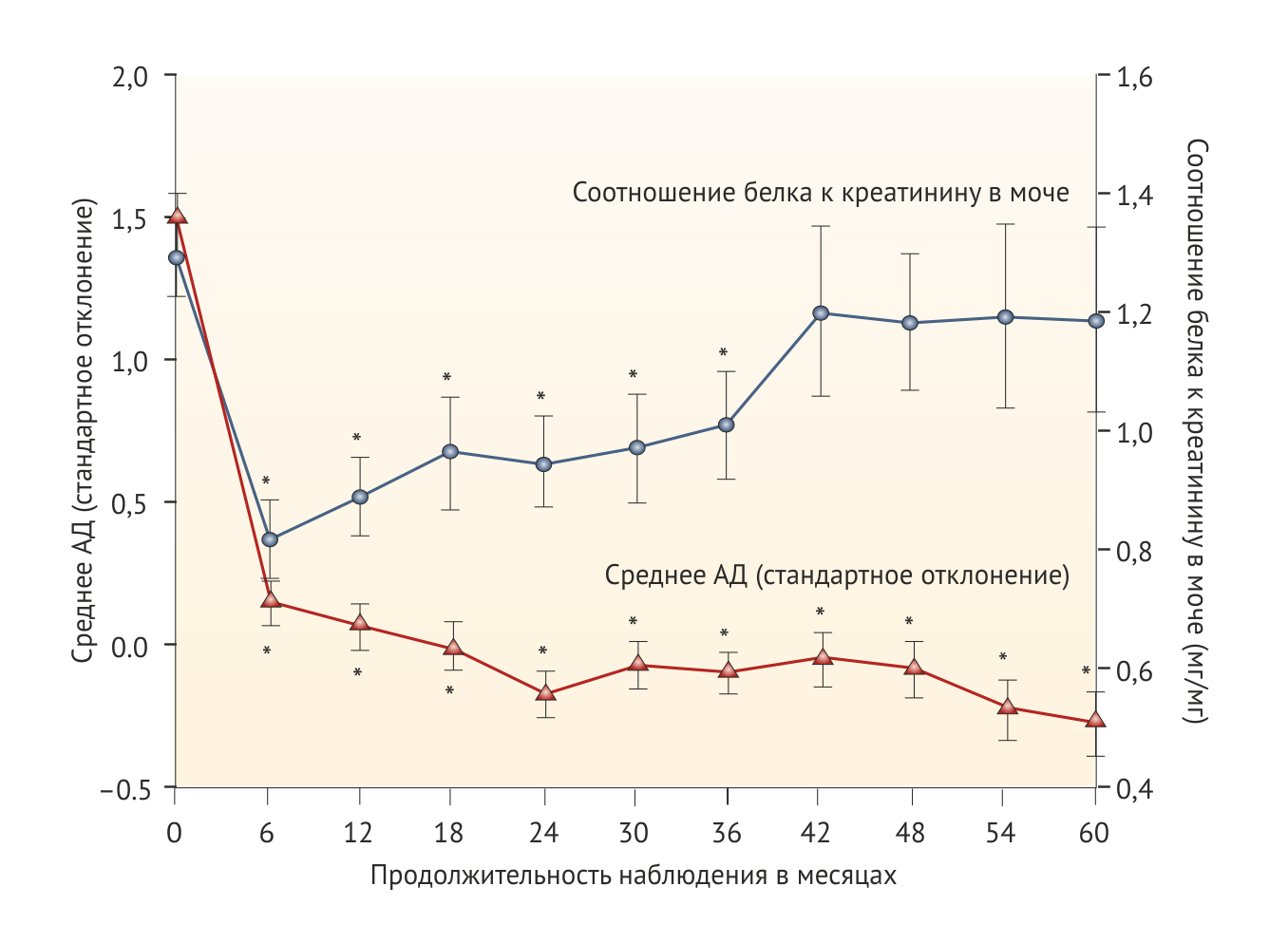
Неясно, существуют ли преимущества от перехода на сартаны или добавления их к терапии при долгосрочном применении иАПФ на фоне продолжающейся протеинурии. В предыдущих исследованиях, возможно, применяли недостаточно высокие дозы иАПФ для подавления внутрипочечных медиаторов РААС и достижения максимального нефропротективного эффекта. В исследовании SMART изучалась возможность достижения большего нефропротективного эффекта использованием доз, превышающих необходимые для достижения оптимального АД. У взрослых пациентов с ХБП и постоянной макропротеинурией исследовали влияние стандартной (16 мг), четырехкратной (64 мг) и восьмикратной суточной дозы (128 мг), в результате наблюдали дозозависимое снижение протеинурии на треть, в то время как на уровень АД, СКФ, уровень калия в крови никакого влияния не наблюдалось [89].
Антипротеинурический ответ в первые 2–3 месяца является хорошим предиктором долговременного нефропротективного эффекта [53] и может полезен для подбора необходимой дозы препарата. Специфическое влияние блокаторов РААС на внутриклубочковую гемодинамику, при старте терапии индуцирует падение СКФ на 10–15 %. Это падение может вызвать повышение уровня креатинина сыворотки у пациентов с ХБП. Тем не менее, это «стартовое» падение СКФ на самом деле является благоприятным предиктором долгосрочного нефропротективного эффекта [90]. Острое ухудшение функции почек вскоре после начала терапии ингибиторами АПФ или сартанами является крайним редким осложнением в результате недиагностированной гиповолемии или двустороннего стеноза почечных артерий.
Антагонисты альдостероновых рецепторов и прямой ингибитор ренина (алискирен) могут использоваться в качестве дополнительных препаратов, ингибирующих РААС. Преимущества применения этих препаратов в дополнение к стандартной терапии ингибиторами АПФ или сартанами были оценены в мета‑анализе 2009 года на более чем 900 пациентах с ХБП [91]. Хотя такая комбинация позволяла достичь максимального уменьшения протеинурии, она значительно увеличивала риск гиперкалиемии. Прямые ингибиторы ренина представляют собой новый класс антигипертензивных препаратов, которые блокируют превращение ангиотензиногена в ангиотензин I. Алискирен, первый одобренный препарат данного класса, снижает АД с эффективностью, аналогичной ингибиторам АПФ и сартанам, а комбинированная терапия дает большее снижение АД, нежели монотерапия [92]. Добавление алискирена к терапии лозартаном оказывает синергетический антипротеинурический эффект у пациентов с диабетической нефропатией [93]. Тем не менее, долгосрочный нефропротективный потенциал от применения ингибиторов ренина и антагонистов альдостеровновых рецепторов еще нужно доказать.
Антигипертензивные препараты других классов.
Существует большое количество антигипертензивных препаратов, не действующих на РААС. БМКК имеют хороший профиль безопасности и неплохо справляются с задачей достижения целевого АД у пациентов с ХБП. Дигидропиридиновые БМКК (нифедипин, амлодипин) не обладают антипротеинурическим эффектом и даже могут способствовать увеличению протеинурии, поэтому в терапии артериальной гипертензии должны использоваться только в комбинации с ингибиторами АПФ [94]. Напротив, недигидропиридиновые БМКК (дилтиазем, верапамил) способствуют снижению протеинурии и, соответственно, имеют нефропротективный эффект.
У пациентов с ХБП часто наблюдается гиперактивация симпатической нервной системы, следовательно, возможно применение адреноблокаторов. Бета-адреноблокаторы хорошо снижают давление у пациентов с ХБП. Примечательно, что в исследовании AASK метопролол обладает антипротеинурическим эффектом, сопоставимым с таковым у рамиприла [51]. Антипротеинурическое действие бета-блокаторов, по-видимому, напрямую связано с их воздействием на симпатическую систему. Новые бета-блокаторы, такие как карведилол, показывают даже большую антипротеинурическую активность по сравнению с классическими представителями этой группы, такими как атенолол [95, 96].
Эндотелин-1 также участвует в развитии и прогрессировании ХБП и сердечно-сосудистых заболеваний. Поэтому селективные антагонисты рецепторов эндотелина-A тоже могут обладать нефропротективным эффектом. В исследовании 2009 года их применение уменьшало протеинурию и артериальную ригидность у 22 пациентов с ХБП без диабета. Частично эти эффекты не зависели от уровня АД [97].
Комбинированная терапия
Поскольку артериальная гипертензия является мультифакториальным заболеванием, монотерапии часто бывает недостаточно для достижения целевого АД. Предпочтительная терапия артериальной гипертензии у больных ХБП должна сочетать в себе ингибиторы РААС, диуретики или БМКК. Комбинированные препараты с фиксированной дозой становятся все популярнее и могут быть полезными в достижении приверженности терапии.
Исследование ACCOMPLISH изучало эффект комбинации сартанов с тиазидным диуретиком или БМКК у более 11 000 пациентов с гипертонией и повышенным риском сердечно-сосудистых и почечных осложнений [98]. Комбинация с БМКК оказалась эффективнее комбинации с диуретиками.
Анализ вторичных конечных точек показал лучший нефропротективный эффект в группе «лозартан-амлодипин» в подгруппе больных ХБП, у которых целевой уровень АД достигался быстрее, чем в контрольной группе. Напротив, снижение протеинурии было более заметным в группе «лозартан-тиазидный диуретик». Подробный анализ относительно влияния уровня АД и протеинурии на состояние функции почек в довольно малой группе пациентов с ХБП и протеинурией, к сожалению, не проводился [97].
Комбинация иАПФ и сартанов в сравнении с монотерапией незначительно влияет на давление и увеличивает антипротеинурический эффект на 30–40 % [66, 99–101]. Тем не менее, несмотря на значительное снижение протеинурии, у пациентов с высоким сердечно-сосудистым риском вследствие атеросклероза и сахарного диабета комбинация иАПФ-сартан приводит к неблагоприятным для почек последствиям (включая и летальный исход), в отличие от монотерапии сартанами или иАПФ [66].
Учитывая, что явление нон-диппинга само по себе является независимым фактором риска прогрессирования ХБП, время приема антигипертензивных препаратов может иметь важное значение. Даже простой прием препаратов на ночь, учитывая их фармакокинетику, может более эффективно снижать уровень ночного АД, тем самым обеспечивая частичное восстановление физиологический ночной динамики АД. Хотя прицельно такая «хронотерапия» не изучалась на пациентах с ХБП, антипротеинурический эффект валсартана был ассоциирован с суточной динамикой: ночное снижение АД в результате вечернего приема препарата заслуживает дальнейшего изучения [102].
Следует отметить, что гипертония и протеинурия — не единственные факторы, способствующие прогрессированию почечной недостаточности. Хотя это выходит за рамки настоящего обзора, уже накоплено достаточно данных, показывающих, что существует довольно много других факторов риска, таких как анемия, метаболический ацидоз, дислипидемия, нарушения минерального обмена, которые независимо влияют на прогрессирование ХБП [103–106].
Заключение
Гипертония и протеинурия являются и маркерами, и патофизиологическими механизмами прогрессирования ХБП. Экспериментальные и клинические данные свидетельствуют о том, что антагонисты РААС помогают сохранить почечную функцию не только посредством снижения уровня АД, но и с помощью специфического антипротеинурического и противовоспалительного эффекта. Вопрос, оказывает ли какое-либо влияние на прогрессирование ХБП строгий контроль АД, остается открытым. По крайней мере у детей с ХБП и пациентов с протеинурией это достоверно оказывает долгосрочный нефропротективный эффект. Следовательно, для этих подгрупп пациентов целевое АД должно быть ниже нормального и препаратами выбора должны быть антагонисты РААС.
