
Гомоцистинурия
Впервые гомоцистинурия была описана в 1962 году в Северной Ирландии: Карсон и Нилл (Carson и Neill) наблюдали двух светловолосых голубоглазых братьев с выраженной задержкой развития и офтальмологическими нарушениями. Спустя десятилетие, в 1972 году, Фриман и его коллеги описали другой вариант заболевания, обусловленный дефицитом фермента метилентетрагидрофолатредуктазы. Авторы обследовали 15-летнюю чернокожую девушку, у которой задержка нервно-психического развития была выраженам слабее, но в течение последних двух лет наблюдались прогрессирующие психические нарушения. Через 12 лет Шух (Schuh) и его соавторы описали случай гомоцистинурии у грудного ребенка с выраженной задержкой психомоторного развития и признаками мегалобластической анемии.
Гомоцистинурия — наследственное нарушение обмена метионина, наследуемое по аутосомно-рецессивному типу. Метионин — незаменимая серосодержащая аминокислота, которая используется для создания непротеиногенной аминокислоты гомоцистеина, синтеза белков (взаимодействие метионина с тРНК является необходимым для создания первой пептидной связи будущего белка) и ряда других важных соединений, благодаря тому, что является донором метильной группы.
В зависимости от того, на каком уровне происходит нарушение обмена метионина, выделяют несколько форм заболевания:
- Классическая гомоцистинурия — обусловлена дефицитом фермента цистатион-бета-синтазы (CbS), лимитирующего синтез цистатиона из гомоцистеина. Ген CBS (кодирующий вышеупомянутый энзим) локализован на длинном плече 21 хромосомы в локусе 1q21-q22.1. Его различные мутации способны привести к развитию гомоцистинурии двух разных фенотипов: пиридоксин (витамин B6) зависимого и пиридоксин (витамин B6) независимого. Средняя частота встречаемости данной формы заболевания до сих пор точно не установлена, т. к. неонатальный скрининг на гомоцистинурию проводится не во всех странах (есть данные, что она составляет 1: 58–1:335000, в странах Ближнего Востока 1:1800–1:8000).
- Гомоцистинурия, обусловленная дефицитом фермента метилентетрагидрофолатредуктазы (MTHFR). Причинный ген картирован на коротком плече 1 хромосомы в локусе 1p36.22. Заболевание встречается крайне редко, его частота не установлена.
- Гомоцистинурия, обусловленная нарушением метаболизма кобаламина (Сbl). Имена эта форма была описана в 1984. Заболевание встречается крайне редко, частота его не установлена. Существует несколько типов гомоцистинурии, связанных с кобаламинами, к настоящему времени наиболее известны:
-
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип G, CblG). Ген локализован на коротком плече 5 хромосомы.
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип Е, CblE). Ген локализован на длинном плече 1 хромосомы.
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип G, CblG). Ген локализован на коротком плече 5 хромосомы.
Патофизиология
При классическом варианте гомоцистинурии из-за дефекта CbS происходит накопление гомоцистеина и его метаболитов в крови и моче. Гомоцистеин синтезируется во всех органах и является токсичным веществом в первую очередь для эндотелия и нейронов. Его токсичность обусловлена, во-первых, наличием реактивной сульфгидрильной группы, которая способна к окислению при физиологическом pH в присутствии кислорода, что ведет к появлению активных форм кислорода и дальнейшему перекисному окислению липидов, повреждению ДНК и апоптозу. Во-вторых, гомоцистеин способен формировать дисульфидные связи со свободной сульфгидрильной группой цистеина в белках, нарушая функцию последних. В-третьих, один из метаболитов гомоцистеина, гомоцистеина тиолактон, соединяясь с лизином протеинов, провоцирует формирование токсических полимеров, агрегатов или амилоида, что является фактором риска нейродегенерации. Помимо этого, белки, присоединившие гомоцистеина тиолактон, становятся антигенном и способствуют развитию иммунной реакции, являющейся важным модулятором атерогенеза.
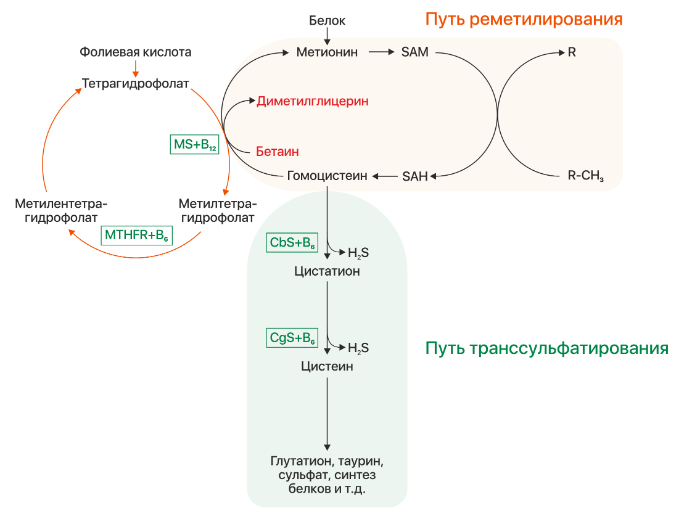 Рисунок 1 | Метаболизм гомоцистеина. CbS — цистатион-бета-синтаза, MS — метионинсинтаза, MTHFR — метилентетрагидрофолатредуктаза, SAM — S-аденозинметионин, SAH — S-аденозингомоцистеин.
Рисунок 1 | Метаболизм гомоцистеина. CbS — цистатион-бета-синтаза, MS — метионинсинтаза, MTHFR — метилентетрагидрофолатредуктаза, SAM — S-аденозинметионин, SAH — S-аденозингомоцистеин.
Несмотря на то, что синтез гомоцистеина происходит практически повсеместно в организме, его утилизация реализуется преимущественно в почках и печени двумя путями: реметилированием и транссульфарацией (см. рис. 1). В сосудистой ткани и коже за неимением ферментов другого пути происходят реакции реметилирования, остальные ткани осуществляют детоксикацию гомоцистеина обоими путями в том или ином соотношении.
При повышение уровня клеточного метионина осуществляется путь транссульфарации: происходит синтез гомоцистеина, который в свою очередь конвертируется в цистатион под действием фермента CbS и кофермента витамина B6. Затем из цистатиона под действием цистатион-гамма-лиазы (CgL) и пиридоксина образуется цистеин. При низком же уровне метионина происходит реакция реметилирования: гомоцистеин принимает метильную группу от метилтетрагидрофолата, превращаясь в метионин. Данная реакция происходит в присутствии фермента метионинсинтазы и кофермента метилкобаламина (витамин B12, CblG), поэтому при нарушении обмена кобаламинов (Cbl) будет развиваться клиника гомоцистинурии. Синтез донора метильной группы регулируется ферментом метилентетрагидрофолатредуктазой (MTHFR), его дефект будет так же вести к развитию заболевания.
Клиническая картина
Клиническая картина гомоцистинурии зависит от формы заболевания и может иметь различную степень выраженности.
При классической гомоцистинурии больные дети имеют мягкие, вьющиеся светлые волосы, румянец на щеках, голубой цвет глаз и «морфаноидный» внешний вид — высокий рост, астеничное телосложение, длинные конечности, арахнодактилия. Помимо этого, возможно развитие кифосколиоза с воронкообразной или килевидной деформацией грудной клетки, вальгусной установки коленных суставов, умеренного остеопороза и переломов у детей старшего возраста. Наиболее типичным поражением глаз являются подвывих (вывих) хрусталика, миопия и вторичная глаукома, также встречаются дети с атрофией зрительных нервов и отслойкой сетчатки. Интеллект детей снижен значительно, задержка развития обращает на себя внимание еще в младенчестве. Неврологическая симптоматика весьма полиморфна и меняется с возрастом (см. табл. 1). Изменения в сердечно-сосудистой системе проявляются развитием тромбоэмболии в сосудах мелкого и среднего калибра, что является главной причиной инсультов и ранней смерти. B6-зависимая гомоцистинурия обычно протекает мягче, чем B6-независимый вариант.
Для больных гомоцистинурией, вызванной дефицитом MTHFR и нарушением обмена Cbl, характерны микроцефалия, умеренное отставание в психомоторном развитии. При отсутствии лечения возможно резкое нарастание нарушений в нервной системе, иногда приводящей к смерти. У больных, заболевание которых в детстве протекало мягко, с минимальной выраженностью клиники, в подростковом и взрослом возрасте происходит резкое ухудшение состояния. При этом изменение неврологической симптоматики происходит трехфазно: после периода нормального развития в раннем детстве (фаза 1), у детей старшего возраста обнаруживается микроцефалия и психомоторные нарушения (фаза 2), затем происходит стремительное нарастание психо-неврологических симптомов (фаза 3), иногда сопровождающееся дыхательной недостаточностью, что может привести к смерти.
Помимо этого, при нарушении обмена Cbl развивается мегалобластическая анемия, что является важным признаком для дифференциальной диагностики.
Таблица 1 | Неврологическая симптоматика в различных возрастных периодах.
| Нарушение CbS |
Нарушение обмена Cbl |
Нарушение MTHFR |
|
| Неонатальный период, первый год жизни |
|||
| Острые неврологические нарушения |
- | + | + |
| Неонатальные приступы |
- | - | + |
| Задержка развития |
+ | + | + |
| Гидроцефалия |
- | +/- | + |
| Нистагм |
- | + | - |
| Раннее и позднее детство |
|||
| Инсульт |
+ | + | + |
| Нервно-психическая задержка развития, обычно трехфазная |
- | + | + |
| Спастический тетрапарез |
- | + | + |
| Психиатрическая симптоматика |
+ | + | + |
| Нистагм |
- | + | - |
| Подростковый возраст, взрослые |
|||
| Нервно-психическая задержка развития |
- | + | + |
| Комбинированная дегенерация спинного мозга |
- | + | + |
| Инсульт |
+ | + | + |
| Миоклония |
- | + | + |
| Необъяснимые острые психиатрические симптомы |
+ | + | + |
| Нистагм |
- | + | - |
Примечания: CbS — цистатион-бета-синтаза, Cbl — кобаламин, MTHFR — метилентетрагидрофолатредуктаза.
Диагностика
В странах, где неонатальный скрининг включает исследование на гомоцистинурию при помощи теста Гатри или тандемной масс-спектрометрии (подобно фенилкетонурии) — диагностика значительно упрощена. Но к сожалению, таких стран немного и Россия не в их числе.
Заподозрить заболевание у новорожденных детей и детей раннего возраста помогают неврологические и офтальмологические нарушения. При этом важно дифференциировать гомоцистинурию от болезни Морфана в виду схожести фенотипических проявлений. Главные отличия: тип наследования (гомоцистинурия наследуется по аутосомно-рецессивному типу, а болезнь Морфана — по аутосомно-доминантному) и нарушение аминокислотного спектра сыворотки крови (при гомоцистинурии). Помимо этого, у детей с болезнью Морфана наблюдаются менее тяжелое поражение глаз, наличие аневризмы аорты, отсутствие интеллектуальных нарушений и остеопороза.
В качестве скринингового метода обнаружения метионина возможно использование качественной реакции с цианиднитропруссидом — при наличие серосодержащих аминокислот моча окрасится в свекольный цвет. Для дифференциальной диагностики между формами заболевания определяют уровень метионина, гомоцистеина и цистеина в плазме крови и моче (см. табл. 2), а также используют молекулярно-генетические методы.
Таблица 2 | Изменения уровней метионина, гомоцистеина и цистеина при различных формах гомоцистинурии.
| Общий гомоцистеин плазмы |
Метионин плазмы |
Цистеин плазмы и мочи |
|
| Нарушение CbS |
↑↑ |
↑ |
↓ |
| Нарушение обмена Cbl |
↑↑ |
↓ |
↑ |
| Нарушение MTHFR |
↑↑ |
↓ / N |
↑ |
Примечания: CbS — цистеин-бета-синтаза, Cbl — кобаламин, MTHFR — метилентетрагидрофолатредуктаза.
При подтверждении диагноза классическая гомоцистинурия (гомоцистинурия, обусловлена дефицитом фермента CbS)” проводят тест с витамином B6 для определения фенотипического варианта заболевания, что важно для последующего лечения.
Методы лечения
Выбор метода лечения зависит от формы заболевания.
При классической B6-зависимой гомоцистинурии рационально назначение пиридоксина, начиная с малых доз (200–250 мг/день для новорожденных и детей раннего возраста, 400–500 мг/день для детей позднего возраста и взрослых). Доза подбирается индивидуально и принимается в течение нескольких недель под контролем маркеров заболевания. Важно ограничить прием больших доз витамина B6 в течение длительного времени, т. к. он начинает оказывать токсический эффект и может вызвать респираторные нарушения, периферическую нейропатию и рабдомиолиз.
Больным с B6-независимой гомоцистинурией показана строгая ограничительная диета: уровень метионина снижается за счет уменьшения потребления белковых продуктов. Для обеспечения физиологической концентрации метионина и цистеина, а также предотвращения белково-энергетической недостаточности, возможно применение аминокислотных смесей с низким содержанием метионина и высоким цистеина.
Бетаин наряду с метилтетрагидрофолатом (см. рис. 1) является донором метильной группы для гомоцистеина. Его прием при B6-резистентной форме гомоцистинурии обеспечивает альтернативный путь синтеза метионина и уменьшает уровень гомоцистеина в крови.
Прием витамина B12, кофактора метионинсинтазы, возможен при всех формах гомоцистинурии. Его натуральная форма (гидроксикобаламин) более эффективна, чем синтетическая (цианкобаламин). При классической гомоцистинурии витамин B12 назначается per os в малых дозах под контролем уровня витамина в плазме крови для предотвращения дефицита кобаламинов. При заболевании, вызванном дефицитом кобаламинов, витамин B12 применяется инъекционно ежедневно.
Назначение фолиевой кислоты (витамин B9) при классической гомоцистинурии (per os 1–5 мг/день ) предотвращает развитие фолатной недостаточности. При дефекте обмена Cbl длительный прием больших доз (5–30 мг/день) витамина B9 компенсирует фолатную ловушку и вызванные ею гематологические изменения (при недостаточности витамина B12 нарушается цикл фолатов и количество фолиевой кислоты в клетке постепенно уменьшается). При недостаточности фермента MTHFR также назначаются большие дозы фолиевой кислоты (5–45 мг/день) и рибофлавин (витамин B2), который является кофактором фермента и может стабилизировать мутантный энзим (см. рис. 1).
Другие методы лечения, обладающие потенциалом:
- снижение pH мочи для увеличения экскреции гомоцистеина тиолактона (подобный метод используется для лечения мочекаменной болезни);
- восстановление функции мутантного фермента CbS при помощи осмолитов. Осмолиты — низкомолекулярные органические соединения, накапливающиеся в клетке, которая находится в стрессовом состоянии, — способны восстанавливать активностьдефектного энзима. Исследования, проводимые на рекомбинантных дрожжах и бактериях, показали эффективность метода, возможно в дальнейшем он будет применяться для лечения классической гомоцистинурии.
Источники:
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Schiff M., Blom H.J. Treatment of Inherited Homocystinurias. // Neuropediatrics 43, 2012: pp 295-304.
- Kumar T. et al. Homocystinuria: Therapeutic approach. // Clinica Chimica Acta, 2016.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] - Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] - М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Гомоцистинурия у детей”, 2016. https://www.pediatr-russia.ru/news/recomend
