
Депрессия. Что мы знаем?

По данным Всемирной организации здравоохранения, депрессия является одним из наиболее распространенных психических расстройств и основной причиной смертности от самоубийств. Примерно 4,4% населения мира страдает от депрессии [1]. Рост глобальных показателей COVID-19 сопровождался увеличением распространенности значительных нейропсихиатрических расстройств, которые могут быть результатом социальных факторов, таких как изоляция и безработица [2,3]. Эта проблема общественного здравоохранения признана глобальной психологической пандемией [4], что требует крупного инвестирования в исследования психического благополучия [5]. Согласно диагностическому и статистическому руководству по психическим расстройствам (DSM-5), большое депрессивное расстройство (MDD) диагностируется при сохранении следующих симптомов в течение как минимум двух недель: депрессивное настроение, чрезмерное чувство вины, ангедония, суицидальные мысли, изменения аппетита и сна, психомоторная отсталость, плохая концентрация и усталость. MDD является сложным и гетерогенным психиатрическим расстройством, и в настоящее время не существует надежных биомаркеров, которые могли бы способствовать объективной диагностике и клинической терапии [4]. Симптомы также включают в себя непреднамеренное изменение веса, бессонницу или гиперсомнию, возбуждение или психомоторную задержку, усталость, чувство бесполезности или вины и попытки самоубийства. Таким образом, MDD значительно снижает качество жизни, что стало серьезной медицинской и социальной проблемой.
Механизмы развития заболевания остаются неясными, так как они имеют разнородную этиологию. Было доказано, что генетика, нейроэндокринология, нейроиммунитет, а также структурные и функциональные расстройства мозга способствуют патофизиологии MDD. Механизмы, которые, как было обнаружено, связаны с депрессией, также включают в себя дисфункциональную гипоталамо-гипофизарно-надпочечниковую ось (HPA) [6], иммуновоспалительные и окислительные пути [7], измененный нервный тонус блуждающего нерва [8] и дисбаланс между нервной волнующей и ингибирующей сигнализацией [9]. К другим факторам, способным привести к развитию депрессии, относят хронический и острый стресс [10], истощение моноаминов [11], плохое питание [12], митохондриальную дисфункцию [13], нарушение метаболизма [14], факторы окружающей среды и эпигенетику [15], инфекцию [16], уровень половых гормонов [17] и т. д. Воспаление в настоящее время считается одной из основных патологий, которые приводят к депрессии. Для объяснения патологии MDD были предложены различные теории, такие как гипотеза макрофагов или теория цитокинов. Гипотеза моноамина также была преобладающей теорией патогенеза депрессии [19].
Депрессия связана с повышенным риском атеросклероза, сердечно-сосудистых заболеваний, гипертонии, инсульта, деменции, нейродегенеративных заболеваний, а также метаболических расстройств, таких как диабет II типа [20]. У людей, страдающих хроническими воспалительными заболеваниями, такими как рак, аутоиммунные заболевания или системные инфекции, также наблюдаются симптомы депрессии [21].
Гипотеза моноаминов
Согласно моноаминовой гипотезе возникновения депрессии, патофизиологической основой депрессии является снижение уровня серотонина, норадреналина и дофамина в центральной нервной системе. Это предположение подтверждается механизмом действия антидепрессантов: было доказано, что все средства, повышающие уровень этих нейромедиаторов в головном мозге, эффективны в облегчении депрессивных симптомов. Считается, что роль моноаминов в формировании отдельных симптомов депрессии неоднородна. Дефицит серотонина может провоцировать появление чрезмерного чувства вины и никчемности, суицидальные идеи, а также нарушение аппетита. При дефиците дофамина и норадреналина — апатию, исполнительную дисфункцию и усталость. Дефицит всех моноаминов в комплексе говорит о подавленном настроении, психомоторной дисфункции и нарушении сна.
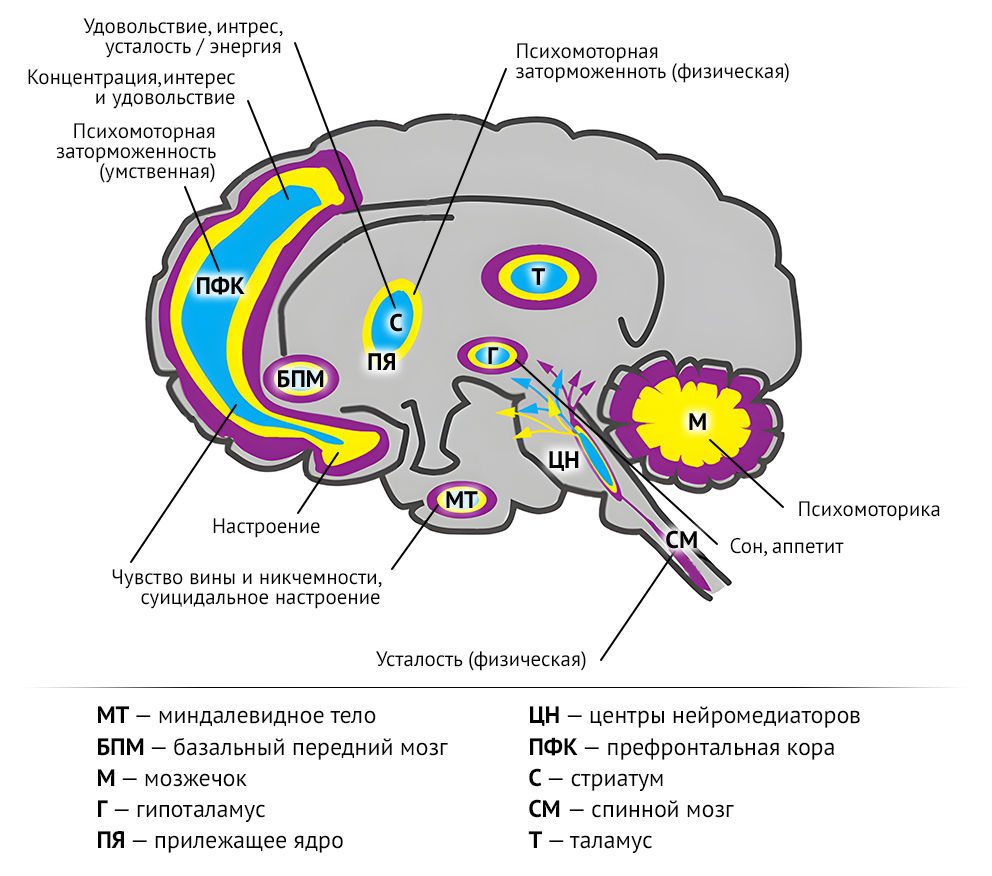
Рисунок 1. Нейротрансмиттеры и их гипотетически неисправные мозговые цепи в областях, связанных с диагностическими симптомами депрессии
Первым поколением антидепрессантов были ингибиторы моноаминоксидазы (МАОИ) и трициклические антидепрессанты, повышающие уровень моноаминов в синаптической щели путем ослабления активности моноаминоксидазы или переносчиков нейромедиаторов соответственно. Для того чтобы минимизировать их серьезные побочные эффекты из-за блокирования определенных постсинаптических рецепторов, были разработаны более специфические и безопасные антидепрессанты. Второе поколение антидепрессантов включало селективные ингибиторы обратного захвата серотонина (СИОЗС): флуоксетин, флувоксамин, пароксетин, сертралин или циталопрам; селективные ингибиторы обратного захвата норадреналина (SNRI): дезипрамин или ребоксетин; двойные ингибиторы обратного захвата серотонина и норадреналина: дулоксетин, венлафаксин и милнаципрам; и многоцелевой антидепрессант вортиоксетин. Были предложены другие соединения с нетипичным механизмом действия, но связанные с активностью моноаминов (например, бупропион, миртазапин или агомелатин).
Для улучшения общих результатов при попытках управлять депрессией в клинической практике были приняты различные фармакологические стратегии, включая увеличение дозы антидепрессанта, смену антидепрессанта, использование комбинаций антидепрессантов и усиленную терапию – эти стратегии улучшают клинический ответ. Одна из ранних стратегий, используемых, когда пациенты неудовлетворительно реагируют на антидепрессанты первого выбора (даже в более высоких дозах), заключается в переходе на другой тип антидепрессанта (например, SNRI, двойной антидепрессант или бупропион) или на другой SSRI. Таким образом, комбинирование антидепрессантов также часто используется для повышения терапевтической эффективности у пациентов, которым не удается достичь желаемого эффекта при начальном лечении (например, СИОЗС и трициклическими антидепрессантами). Помимо повышения эффективности антидепрессантов, такие комбинированные стратегии также используются для повышения переносимости. Например, бупропион противодействует сексуальной дисфункции, вызванной СИОЗС. Стратегия усиления обычно подразумевает использование дополнительного препарата, который не является антидепрессантом, для повышения эффективности моноаминергических препаратов и / или для сокращения времени, необходимого им для достижения клинического эффекта. Основываясь на имеющейся литературе, заслуживают определенного внимания следующие методы усиления:
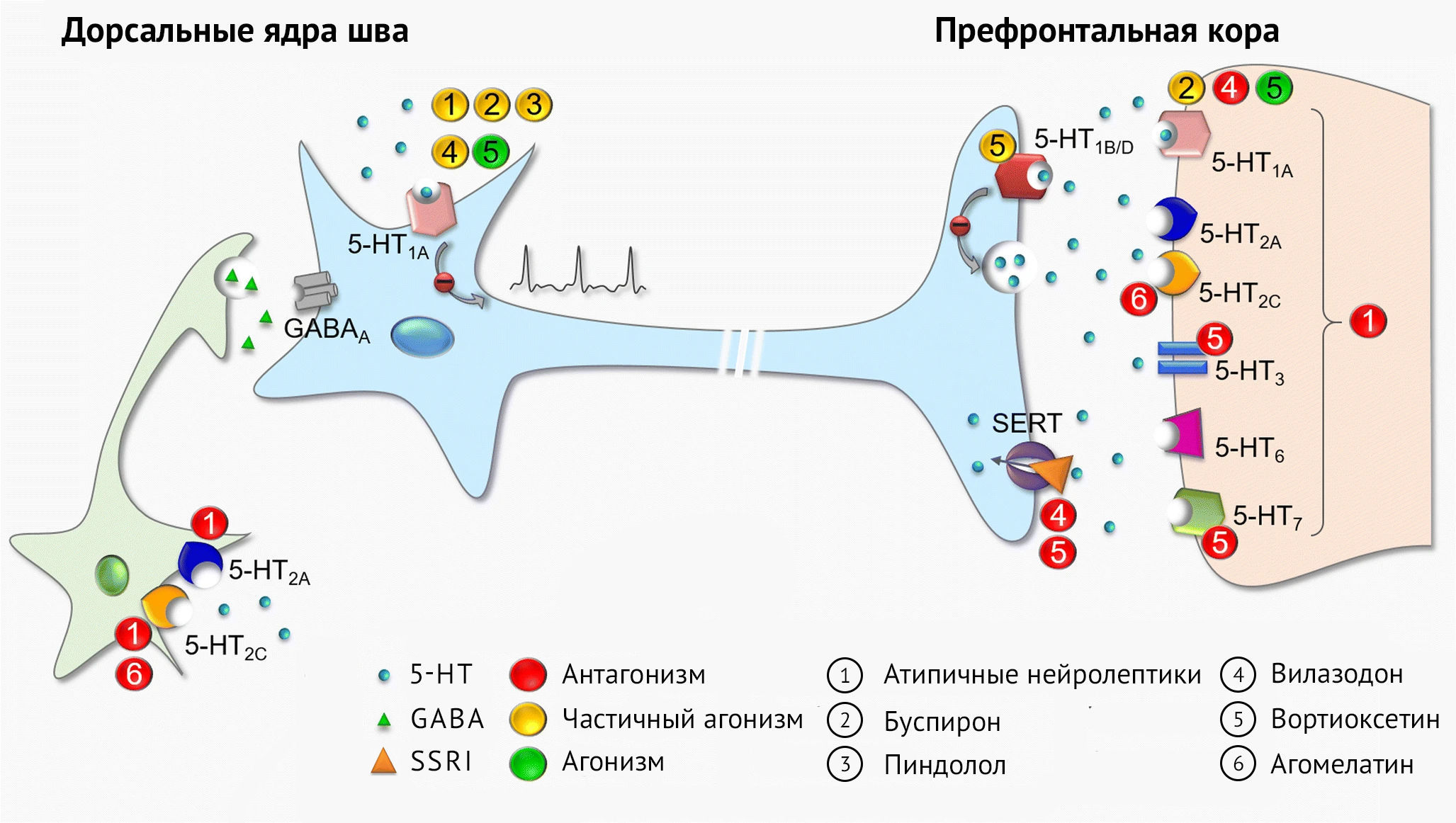
Рисунок 2. Потенциальные механизмы, связанные с усиливающей терапией и мультицелевыми антидепрессантами
На этой схеме показаны множественные потенциальные мишени на серотонинергических и гамкергических нейронах в дорсальном ядре шва или на нейронах коры головного мозга для усиливающих агентов и многозадачных антидепрессантов, а также указаны их фармакологические эффекты (антагонизм, частичный агонизм или агонизирующее действие). Стратегии усиления (атипичные нейролептики (1), буспирон (2) и пиндолол (3)) основаны на сочетании ингибирования SERT, обычно с использованием селективных ингибиторов обратного захвата серотонина (СИОЗС), с другими средствами, которые действуют непосредственно на серотонинергические рецепторы. Хотя атипичные нейролептики обладают отчетливым сродством к различным рецепторам моноаминергических нейромедиаторов, этот рисунок иллюстрирует только наиболее общие эффекты на серотонинергические рецепторы. Буспирон и пиндолол действуют на рецепторы 5-HT1A, хотя следует отметить, что пиндолол оказывает преимущественное действие на пресинаптические, а не на постсинаптические рецепторы 5-HT1A. Многофункциональные антидепрессанты вилазодон (4) и вортиоксетин (5) ингибируют обратный захват серотонина в дополнение к их сродству к серотонинергическим рецепторам. Напротив, агомелатин (6) обладает смешанным механизмом действия, нацеленным на рецепторы 5-HT2C и мелатонинергическую систему.
Теория цитокинов
Стрессовые события активируют симпатическую нервную систему и гипоталамо-гипофизарно-надпочечниковую ось (HPA), в следствие чего происходит выделение химических медиаторов, защищающих организм от стресса. Изменения в оси HPA и симпатической нервной системе в основном влияют на иммунную систему. Хроническое воспаление является важнейшим компонентом хронических заболеваний, включая депрессию. Кроме того, иммунный дисбаланс, особенно отражающийся в снижении активности естественных клеток-киллеров, был связан с подавленным настроением. Как экспериментальные, так и клинические исследования показали, что повышение уровня провоспалительных цитокинов и гормонов стресса, таких как глюкокортикоиды, в значительной степени способствует поведенческим изменениям, связанным с депрессией. Данные свидетельствуют о том, что воспаление играет ключевую роль в патологии заболеваний, связанных со стрессом, однако эта связь еще не полностью изучена.
Секреция гипоталамусом кортикотропин-рилизинг гормонов подавляет иммунные реакции, опосредуя высвобождение глюкокортикоидов из надпочечников. Глюкокортикоиды подавляют цитотоксичность и пролиферацию лимфоцитов [22,23]. Кроме того, они снижают экспрессию нескольких провоспалительных цитокинов, таких как интерлейкин (IL)-6 и фактор некроза опухоли (TNF)-α. При этом глюкокортикоиды повышают экспрессию таких противовоспалительных цитокинов как IL-10 и TNF-β [24,25]. Глюкокортикоиды также оказывают провоспалительное действие на иммунную систему [26, 27]. Они усиливают функцию инфламмасомы NLRP3 за счет увеличения секреции IL-1β в ответ на аденозинтрифосфат. Инфламмасомы представляют собой множественные белковые комплексы, которые воспринимают внешние и внутренние опасные сигналы, приводящие к расщеплению провоспалительных цитокинов до зрелых цитокинов, включая IL-1β и IL-18. Провоспалительные факторы, включая IL-1, IL-6 и TNF-α, активируют гипофизарно-надпочечниковую систему, что приводит к повышению сывороточных уровней адренокортикотропного гормона (АКТГ) и глюкокортикоидов, которые, в свою очередь, подавляют выработку провоспалительных факторов. [28,29]. Иммунная система и ось HPA взаимодействуют, формируя механизм отрицательной обратной связи. Петли отрицательной обратной связи могут нарушаться из-за снижения экспрессии цитоплазматических глюкокортикоидных рецепторов (GRS) и GR-управляемых противовоспалительных генов, что приводит к снижению чувствительности к глюкокортикоидам, когда стрессоры чрезмерно стимулируют цитокины.
Провоспалительные цитокины, активирующие микроглию, приводят к периферическому накоплению моноцитов и макрофагов. Они также были обнаружены в головном мозге после воздействия стресса [30,31]. Макрофаги головного мозга, составляющие микроглию, считаются основным источником провоспалительных цитокинов. Микроглия экспрессирует как глюкокортикоидные, так и минералокортикоидные рецепторы и напрямую реагирует на пик уровня кортикостерона. Кроме того, GR высоко сконцентрированы в префронтальной коре и гиппокампе. Таким образом, связанный со стрессом кортикостерон косвенно влияет на микроглию. Активированная микроглия демонстрирует изменение морфологии, которое наблюдается в виде гипертрофированной сомы с увеличенной ветвью. Это приводит к увеличению количества цитокинов, которые рекрутируют моноциты с периферии. Увеличение количества макрофагов и периферических моноцитов способствует выработке в головном мозге провоспалительных цитокинов, таких как IL-1β, TNFα и IL-6 [32].
Стрессовые события являются основополагающими в провоцировании эпизодов тяжелого депрессивного расстройства (MDD). Пациенты с депрессией предрасположены к активации оси HPA и гиперкортизолемии, повышению уровня гормонов стресса и кортикотропин-рилизинг-гормона, а также увеличению уровня АКТГ. MAPK повышают активность мембранных транспортеров серотонина, а серотонин является наиболее важным нейромедиатором, связанным с депрессией [33]. Основным принципом воспалительной депрессии является активация иммунного ответа, в частности выработки цитокинов. Это влияет на уровни нейрохимикатов, которые приводят к MDD. Стресс может способствовать развитию депрессивноподобного поведения, способствуя экспрессии воспалительных цитокинов. Недавно новый кинурениновый путь (КП) привлек внимание в теории цитокинов. Провоспалительные цитокины активируют КП, влияя на метаболизм триптофана и секретируя нейротоксины, которые могут либо снижать выработку серотонина, либо способствовать обратному захвату серотонина [34].
Повышенные уровни медиаторов воспаления, включая цитокины, белки острой фазы, хемокины, молекулы адгезии и простагландины, также были обнаружены в ЦНС, крови и спинномозговой жидкости пациентов с депрессией. Воздействие хронического стресса в течение четырех недель значительно активирует воспалительные цитокины, включая IL-18, IL-1β, TNF-α и индуцируемую воспалением синтазу оксида азота (NOS). Повышение уровня провоспалительных цитокинов провоцирует депрессивноподобное поведение. Блокирование воспалительных цитокинов или индуцируемых NOS такими препаратами, как миноциклин, помогает устранить депрессивноподобное поведение, вызванное стрессом. Было показано, что некоторые антидепрессанты обладают противовоспалительным действием. Антидепрессанты и нестероидные противовоспалительные препараты, такие как миноциклин, смягчают активацию микроглии и снижают уровень IL-6 в крови и секрецию центральных цитокинов, тем самым уменьшая изменения в поведении.
Инфламмасомы представляют собой множественные белковые комплексы, которые стимулируют выработку и созревание провоспалительных факторов, таких как IL-18 и IL-1β, для индукции врожденной иммунной защиты. Инфламмасома NLRP3 была вовлечена в индукцию депрессивноподобного поведения в мышиной модели, индуцированной липополисахаридом (LPS). Недавнее исследование продемонстрировало, что защитная роль ингибирования каспазы-1 в функционировании мозга и кишечной микробиоте провоцирует депрессивное и тревожноподобное поведение.
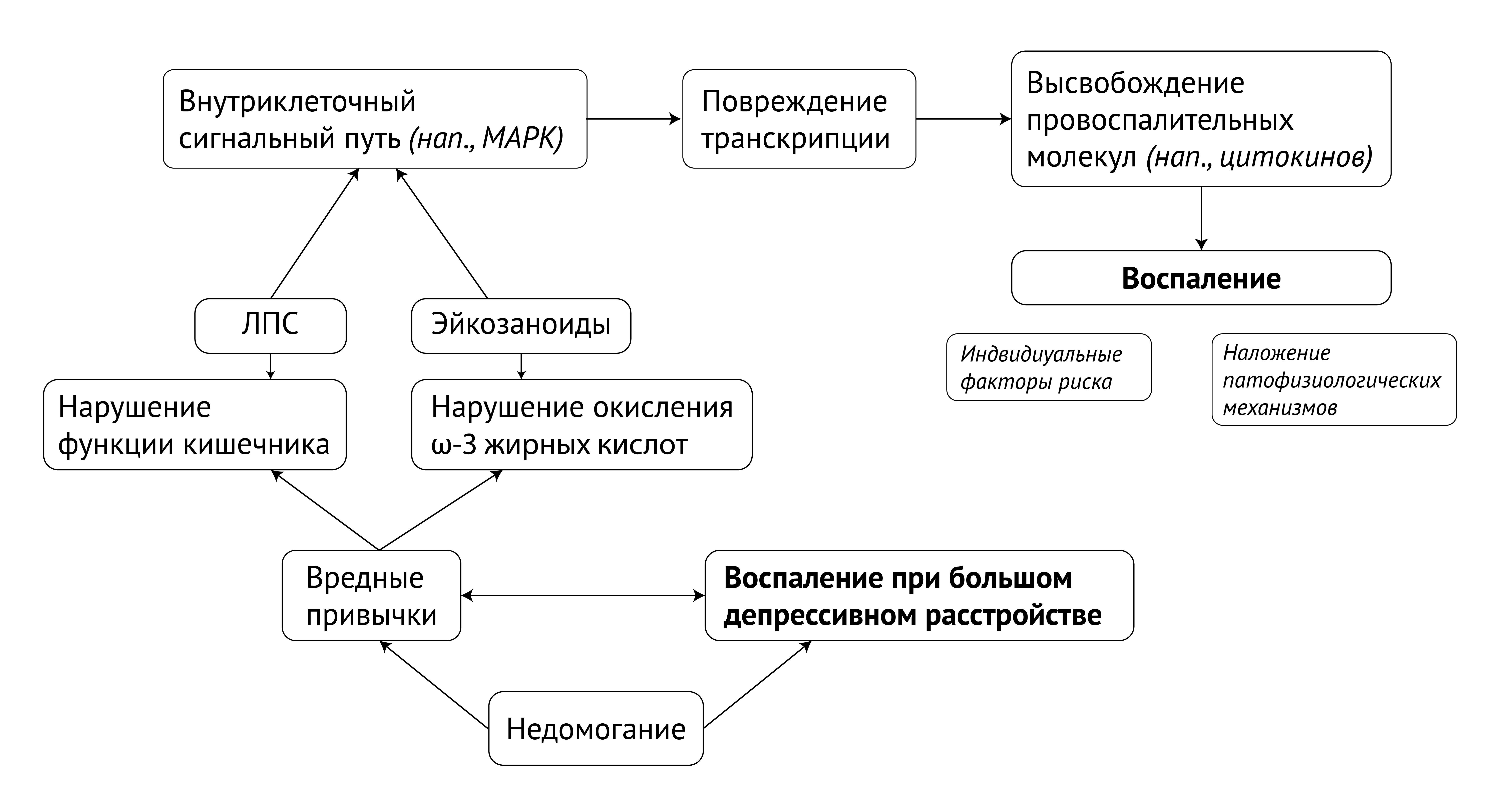
Рисунок 3. Модель реципрокных взаимодействий между воспалительными изменениями низкой степени выраженности и MDD
Хроническое воспаление и депрессия
MDD связан с заболеваниями, характеризующимися активированной иммунной системой, такими как аутоиммунные заболевания (системная красная волчанка (СКВ), сахарный диабет 1 типа и ревматоидный артрит (РА)), аллергии и инфекции (сепсис). У пациентов как с астмой, так и с атопией частота МДД примерно на 50% выше. Примерно 36% пациентов с астмой страдают МДД, и уровни их TNFα значительно повышены, в то время как уровни IFNy значительно снижены.
Метаанализы показали, что частота MDD у пациентов с сахарным диабетом почти в два раза выше, чем у пациентов без диабета. Активация воспаления связана с патогенезом диабета, при этом иммунный ответ задействован как при диабете I типа, так и II типа. Маркеры воспаления, включая CRP, IL-1β, IL-1RA и MCP-1, значительно повышены у пациентов с MDD и сахарным диабетом 2 типа.
MDD также чаще встречается у пациентов с ревматоидным артритом (РА). Исследования показали, что риск развития РА у пациентов с MDD на 74% выше, чем у здоровых людей контрольной группы. Кроме того, 17% пациентов с РА имеют MDD. Более 70% пациентов с РА испытывают клинически значимую усталость [35].
Сообщалось о связи между активацией иммунитета и MDD не только при нарушениях, связанных с иммунитетом, но и в случаях инфекций. Сепсис приводит к широким провоспалительным реакциям, которые запускают системный иммунный ответ на инфекционный агент. У пациентов с сепсисом наблюдается существенно повышенный уровень маркеров воспаления. У выживших после сепсиса частота MDD повышена по сравнению с нормальной контрольной группой; однако было обнаружено, что она ненамного выше, чем частота MDD, предшествовавшая инфекции. Такой высокий уровень MDD у пациентов с сепсисом также отражает то, что психологический стресс увеличивает MDD и активацию иммунитета и связан с более высоким риском развития сепсиса. В нескольких исследованиях сообщалось о постсептическом MDD у пациентов-людей, в то время как в исследованиях на животных сообщалось о состояниях сепсиса, приводящих к изменениям эмоций. Исследования на животных также показали, что подавление иммунитета путем подавления пути NF-kB снижает поведение, подобное MDD, на животных моделях. Возможно, существует роль “инициирования” иммунного ответа такими состояниями, как сепсис, что, в свою очередь, вызывает более высокий риск развития MDD.
Микробиом кишечника и депрессия
Известные механизмы возникновения, развития и поддержания депрессивного состояния являются многофакторными и определяются полимодальными изменениями в метаболической, иммунной, эндокринной, желудочно-кишечной и центральной нервной системах (ЦНС). Желудочно-кишечный тракт с его микробиотой, взаимодействующей с внешними сигналами окружающей среды, стрессом, питательными веществами, внутренними системами, включая мозг, также может быть вовлечен в развитие депрессивных состояний. Существует достаточно доказательств связи между кишечной микробиотой и патофизиологией депрессии [36]. Различные стрессы (социальный или эмоциональный, химический, физический, плохое питание и т. д.) могут изменить таксономический состав бактериальных сообществ в кишечнике и, как следствие, приводят к изменениям в различных метаболических путях. Это, в свою очередь, приводит к систематическому воспалительному процессу, который охватывает другие системы органов человека. На текущем уровне исследований наиболее важным моментом является выявление различных биомаркеров метаболитов, которые коррелируют с депрессивными состояниями. Метаболический синдром хорошо задокументирован у пациентов с MDD и в 1,5 раза выше, чем у недепрессивной популяции [37].
Ось кишечник-мозг
Исследования показывают, как изменения в составе микробиоты кишечника влияют на все аспекты физиологии, включая функции мозга и даже поведение. С точки зрения неврологии и психиатрических заболеваний, эта область все еще находится на начальной стадии изучения, но появляется все больше доказательств того, что она играет значительную роль. Факторы, формирующие бактериальный профиль, включают рождение путем кесарева сечения, отсутствие грудного вскармливания, окружающую среду, гестационный возраст, генетику хозяина, подверженность инфекциям (как материнским, так и младенческим) и использование антибиотиков. Кроме того, стресс, особенно в раннем возрасте и в пренатальный период, может оказывать заметное влияние на состав микробиоты, значительно изменяя микробиомный профиль [38]. Используются различные пути для изучения роли двунаправленной связи между кишечником человека и мозгом, так называемой оси «микробиота-кишечник–мозг», у здоровых людей и пациентов, переносящих определенное заболевание.
Бактерии кишечной микробиоты способны продуцировать нейромедиаторы и другие нейрометаболиты, которые связаны с депрессией. Эти бактериальные продукты могут участвовать в стимуляции центральных рецепторов, в периферической стимуляции нервных, иммунных и эндокринных медиаторов, а также участвовать в эпигенетической регуляции ацетилирования гистонов и метилирования ДНК. Нейроны кишечной нервной системы (ENS) непосредственно взаимодействуют с нейрохимическими веществами, вырабатываемыми кишечной микробиотой, тем самым влияя на передачу сигналов в центральную нервную систему (ЦНС). Нарушение действия различных факторов, посредством которых кишечные микроорганизмы воздействуют на различные системы и пути в организме хозяина, обычно демонстрируется во время депрессии. Эти факторы включают воспаление и окислительный стресс, метаболизм триптофана и кинурениновый путь, митохондриальную дисфункцию, нейротрансмиттеры, пластичность мозга и нейротрофические факторы метаболических процессов.
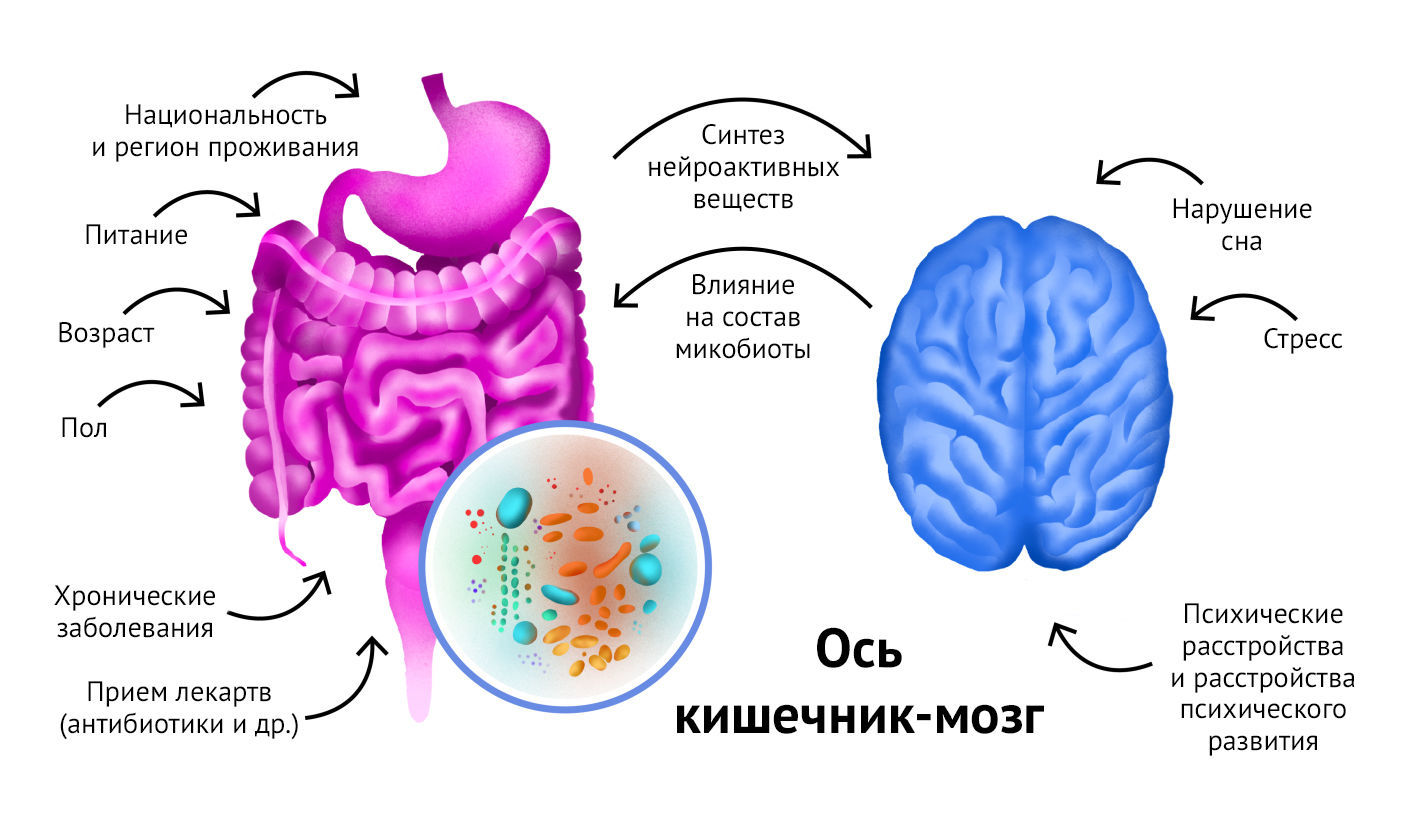
Рисунок 4. Иллюстрация работы системы “ось кишечник – мозг”
Основные достижения
Многочисленные исследования кишечной микробиоты пациентов с депрессией показали дисбиотические различия, включая снижение количества полезных бактерий, продуцирующих бутират: Faecalibacterium prausnitzii, Roseburia intestinalis, родов Ruminococcus, Coprococcus и Dialister, и увеличение количества провоспалительных условно-патогенных бактерий из типов Proteobacteria и Bacteroidetes. Дисбактериоз кишечника (GD) влияет на защитные свойства гематоэнцефалического барьера (ГЭБ), включая модуляцию проницаемости, и может привести к воспалению в головном мозге, что вызывает изменения в настроении и поведении. Патогенный эффект кишечной микробиоты во многих клинических исследованиях был связан с депрессивным и тревожным поведением. Изменения в популяции определенных видов кишечной микробиоты способствуют депрессии, и, с другой стороны, депрессивное состояние может вызвать изменения в микробиоте кишечника, которые приведут к более тяжелой форме депрессии.
Идентификация кишечных бактерий и их метаболитов, которые могут прямо или косвенно влиять на развитие депрессии, имеет большое значение как для создания систем диагностики заболевания, так и для выбора стратегий, направленных на восстановление нормального функционирования микробиоты и, таким образом, на восстановление психического здоровья. Поиск подходов к восстановлению микробиоты при депрессии стал особо актуальным в последнее десятилетие. Подтверждается, что применение пробиотических препаратов, влияющих на высшие функции мозга, положительно влияет на пациентов с депрессивным поведением. Появляется все больше литературы, в которой рассматриваются различные подходы, альтернативные фармацевтическим препаратам, используемым в качестве антидепрессантов. Эти подходы включают диету и использование пищевых добавок, таких как пробиотики, пребиотики, постбиотики. Клинические улучшения уже были продемонстрированы у пациентов с неврологическими расстройствами после использования такого терапевтического подхода.
Кишечные бактерии участвуют во взаимодействии с ЦНС, ЭНС и вегетативной нервной системой (ВНС) по нейроиммунным и нейроэндокринным путям посредством нервных сигналов, передаваемых блуждающим нервом. Иммуномодуляция слизистой оболочки осуществляется микробиотой и ее продуктами, а также химическими сигналами, синтезируемыми микробиотой. Кроме того, кишечная микробиота может контролировать как ЦНС, так и ЭНС посредством выработки и экспрессии нейромедиаторов и нейротрофических факторов. Они модулируют сенсорные афференты кишечника и выработку метаболитов, а также поддерживают целостность кишечного барьера и плотных соединений. Микробиота кишечника участвует в созревании микроглии, нейрогенезе и регуляции формы экспрессии рецепторов нейромедиаторов в головном мозге, регулирует проницаемость ГЭБ. Посредством всех этих механизмов микробиом кишечника человека может быть вовлечен в патогенез клинической депрессии. С другой стороны, воздействие депрессии на микроорганизмы кишечника, регулируемое стрессом, может изменять высвобождение нейромедиаторов и других сигнальных молекул в кишечнике и влиять на нарушение регуляции иммунного ответа.
Понимание нейроиммунных причин депрессии и других психических расстройств, связанных со стрессом, растет, хотя основные механизмы, связывающие иммунную, эндокринную и нервную системы с поведенческими и психологическими симптомами, до конца не изучены. Известным фактором риска развития MDD является активация системы воспалительного ответа. Как правило, основой воспаления являются молекулярные структуры патогенов, включая бактериальные липополисахариды (ЛПС), липопротеины, флагеллины и пептидогликаны, а также эндотоксин. Исследования на животных показали, что периферическое введение ЛПС приводит к аналогичному депрессивному поведению и повышенной экспрессии IL-1β, TNF-α, синтазы оксида азота (iNOS) в гиппокампе и коре головного мозга. Другой тип воспаления рассматривается как “стерильное воспаление”, вызванное психологическим стрессом или молекулярными паттернами, связанными с опасностью/повреждением (DAMPs), иначе определяемыми как сигналы тревоги. Эти биомолекулы высвобождаются в результате повреждения тканей. GD может провоцировать аутоиммунные заболевания из-за неадекватной посттрансляционной модификации белков хозяина. Кишечные микробы экспрессируют широкий спектр ферментов, которые участвуют в посттрансляционной модификации белков (PTMP) в желудочно-кишечном тракте. Во всех случаях бактерии являются источником активации рецепторов (PRRs). Например, активация TLR4 бактериальными LPS запускает так называемые инфламмасомы - рецепторы врожденной иммунной системы, которые дополнительно активируют внутриклеточные провоспалительные каспазы, что приводит к высвобождению провоспалительных цитокинов. Цитокины вызывают депрессивные симптомы, влияя на различные процессы, связанные с эмоциями. Повышенные воспалительные сигналы могут вызывать нарушения метаболизма нейромедиаторов, дестабилизировать состояние нервов, а также нарушать мозговую регуляцию и сигнальные механизмы в поведении и эмоциях.
Таблица 1. Каталог генов ключевых бактериальных ферментов, имеющих отношение к депрессии [39]
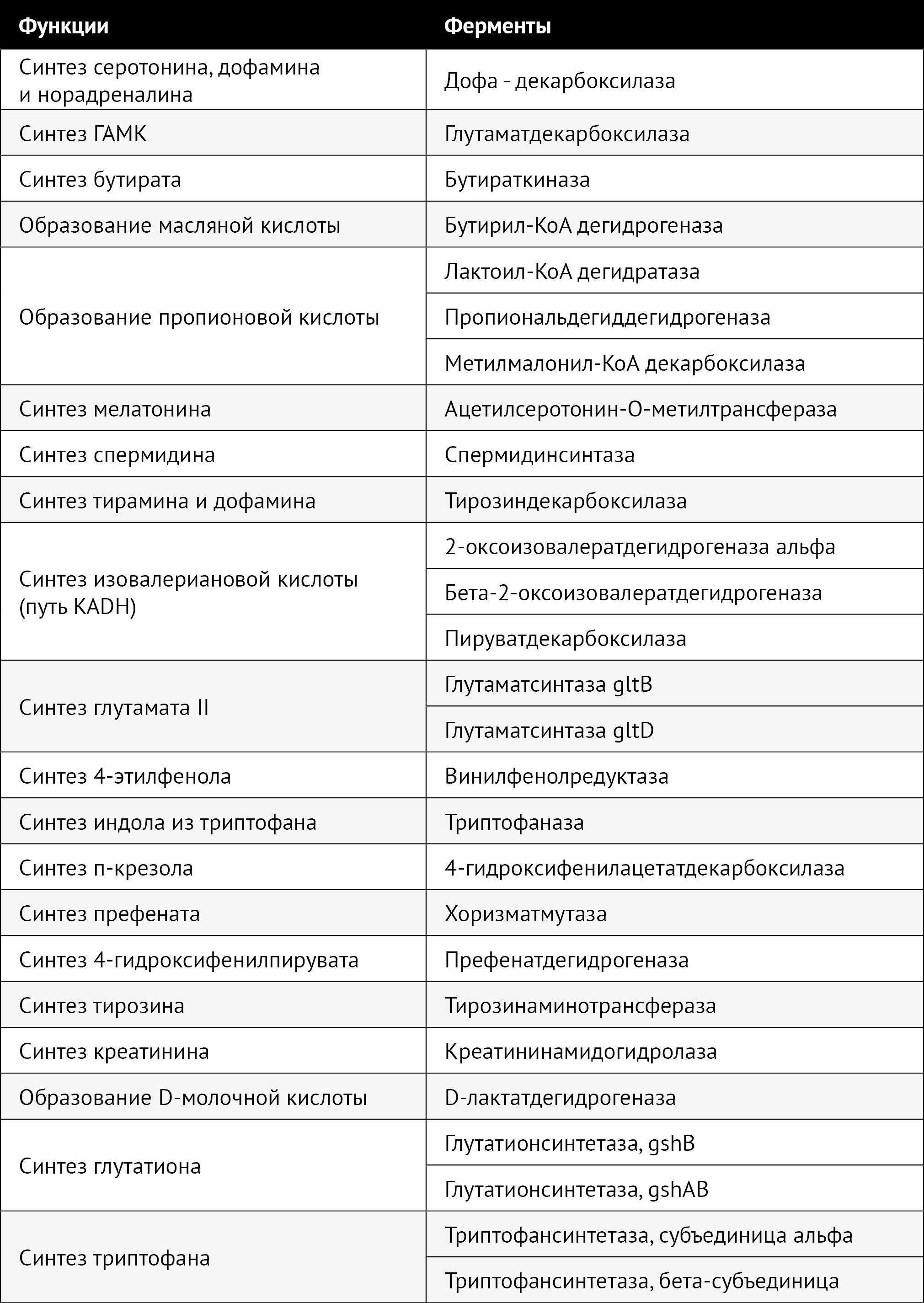
Использование биомаркеров кишечной микробиоты, отражающих нейромодуляторный, иммуномодулирующий и антиоксидантный статус организма-хозяина, при анализе метагеномных данных пациентов с нервно-психическими заболеваниями приобретает все большее распространение. Создание каталогов генов, содержащих функциональные гены, позволит целенаправленно анализировать метагеномные данные. Определение метагеномной сигнатуры в норме имеет решающее значение для выделения генов с диагностическим потенциалом в контексте депрессии. Кодируемые бактериальными генами ферменты, которые участвуют в метаболизме биомаркеров, описанных в этом обзоре, скорее всего, определяют нейро- и иммуномодулирующий потенциал микробиомов. Они способствуют проявлениям депрессии и могут быть использованы для идентификации метаболической сигнатуры кишечной микробиоты пациентов с депрессией.
Список литературы
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017.
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783.
- The Lancet Global Health. Mental health matters. Lancet Glob. Health 2020, 8, e1352.
- Thakur, V.; Jain, A. COVID 2019-suicides: A global psychological pandemic. Brain Behav. Immun. 2020, 88, 952–953.
- Santomauro, D.F.; Mantilla Herrera, A.M.; Shadid, J.; Zheng, P.; Ashbaugh, C.; Pigott, D.M.; Abbafati, C.; Adolph, C.; Amlag, J.O.; Aravkin, A.Y.; et al. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712.
- Burke, H.M.; Davis, M.C.; Otte, C.; Mohr, D.C. Depression and cortisol responses to psychological stress: A meta-analysis. Psychoneuroendocrinology 2005, 30, 846–856.
- Köhler, C.; Freitas, T.; Maes, M.D.; De Andrade, N.; Liu, C.; Fernandes, B.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387.
- Chambers, A.S.; Allen, J.J. Vagal tone as an indicator of treatment response in major depression. Psychophysiology 2002, 39, 861–864.
- Ghosal, S.; Hare, B.D.; Duman, R.S. Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr. Opin. Beha. Sci. 2017, 14, 1–8.
- Kim, I.-B.; Lee, J.-H.; Park, S.-C. The relationship between stress, inflammation, and depression. Biomedicines 2022, 10, 1929.
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The serotonin theory of depression: A systematic umbrella review of the evidence. Mol. Psychiatry 2022.
- Bruce-Keller, A.J.; Salbaum, J.M.; Berthoud, H.-R. Harnessing gut microbes for mental health: Getting from here to there. Biol. Psychiatry 2018, 83, 214–223.
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial impairment: A common motif in neuropsychiatric presentation? The link to the tryptophan-kynurenine metabolic system. Cells 2022, 11, 2607.
- Gheshlagh, R.G.; Parizad, N.; Sayehmiri, K. The relationship between depression and metabolic syndrome: Systematic review and meta-analysis study. Iran. Red Crescent Med. J. 2016, 18, e26523.
- Ortega, M.A.; Fraile-Martínez, Ó.; García-Montero, C.; Alvarez-Mon, M.A.; Lahera, G.; Monserrat, J.; Llavero-Valero, M.; Mora, F.; Rodríguez-Jiménez, R.; Fernandez-Rojo, S.; et al. Nutrition, epigenetics, and major depressive disorder: Understanding the connection. Front. Nutr. 2022, 9, 867150.
- Brasso, C.; Bellino, S.; Blua, C.; Bozzatello, P.; Rocca, P. The impact of SARS-CoV-2 infection on youth mental health: A narrative review. Biomedicines 2022, 10, 772.
- Slavich, G.M.; Sacher, J. Stress, sex hormones, inflammation, and major depressive disorder: Extending Social Signal Transduction Theory of Depression to account for sex differences in mood disorders. Psychopharmacology 2019, 236, 3063–3079.
- Kucukkarapinar, M.; Yay-Pence, A.; Yildiz, Y.; Buyukkoruk, M.; Yaz-Aydin, G.; Deveci-Bulut, T.S.; Gulbahar, O.; Senol, E.; Candansaya, S. Psychological outcomes of COVID 19 survivors at sixth months after diagnose: The role of kynurenine pathway metabolites in depression, anxiety, and stress. J. Neural Transm. 2022, 129, 1077–1089.
- Holtzheimer III, P.E.; Nemeroff, C.B. Future prospects in depression research. Dialogues Clin. Neurosci. 2006, 8, 175–189.
- Friedrich, M.J. Depression is the leading cause of disability around the world. JAMA 2017, 317, 1517.
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56.
- Gallelli, L.; D’Agostino, B.; Vatrella, A.; Fratto, D.; Renda, T.; Galderisi, U.; Piegari, E.; Crimi, N.; Rossi, F.; Caputi, M. Effects of TGF-beta and glucocorticoids on map kinase phosphorylation, IL-6/IL-11 secretion and cell proliferation in primary cultures of human lung fibroblasts. J. Cell. Physiol. 2007, 210, 489–497.
- Gallelli, L.; Pelaia, G.; Fratto, D.; Muto, V.; Falcone, D.; Vatrella, A.; Curto, L.; Renda, T.; Busceti, M.; Liberto, M. Effects of budesonide on p38 MAPK activation, apoptosis and IL-8 secretion, induced by TNF-α and Haemophilus influenzae in human bronchial epithelial cells. Int. J. Immunopathol. Pharmacol. 2010, 23, 471–479.
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The stressed CNS: When glucocorticoids aggravate inflammation. Neuron 2009, 64, 33–39.
- Hill, A.R.; Spencer-Segal, J.L. Glucocorticoids and the brain after critical illness. Endocrinology 2021, 162, bqaa242.
- Elenkov, I.J. Neurohormonal-cytokine interactions: Implications for inflammation, common human diseases and well-being. Neurochem. Int. 2008, 52, 40–51.
- van den Heuvel, L.L.; Suliman, S.; Bröcker, E.; Kilian, S.; Stalder, T.; Kirschbaum, C.; Seedat, S. The association between hair cortisol levels, inflammation and cognitive functioning in females. Psychoneuroendocrinology 2022, 136, 105619.
- Alley, D.E.; Seeman, T.E.; Kim, J.K.; Karlamangla, A.; Hu, P.; Crimmins, E.M. Socioeconomic status and C-reactive protein levels in the US population: NHANES IV. Brain Behav. Immun. 2006, 20, 498–504.
- Danese, A.; Pariante, C.M.; Caspi, A.; Taylor, A.; Poulton, R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc. Natl. Acad. Sci. USA 2007, 104, 1319–1324.
- Johnson, J.; Campisi, J.; Sharkey, C.; Kennedy, S.; Nickerson, M.; Greenwood, B.; Fleshner, M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 2005, 135, 1295–1307.
- Johnson, J.D.; Barnard, D.F.; Kulp, A.C.; Mehta, D.M. Neuroendocrine regulation of brain cytokines after psychological stress. J. Endocr. Soc. 2019, 3, 1302–1320.
- Wohleb, E.S.; Delpech, J.-C. Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog. Neuropsychopharmacol. Biol. Psychiatr. 2017, 79, 40–48.
- Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and therapeutic applications of tumor necrosis factor-α (TNF-α) in major depressive disorder: A systematic review. Int. J. Mol. Sci. 2016, 17, 733.
- Miura, H.; Ozaki, N.; Sawada, M.; Isobe, K.; Ohta, T.; Nagatsu, T. A link between stress and depression: Shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress 2008, 11, 198–209.
- Stebbings, S.; Treharne, G.J. Fatigue in rheumatic disease: An overview. Int. J. Clin. Rheumatol. 2010, 5, 487–502.
- Karl, J.P.; Hatch, A.M.; Arcidiacono, S.M.; Pearce, S.C.; Pantoja-Feliciano, I.G.; Doherty, L.A.; Soares, J.W. Effects of psychological, environmental and physical stressors on the gut microbiota. Front. Microbiol. 2018, 9, 2013.
- Gheshlagh, R.G.; Parizad, N.; Sayehmiri, K. The relationship between depression and metabolic syndrome: Systematic review and meta-analysis study. Iran. Red Crescent Med. J. 2016, 18, e26523.
- Borre, Y.E.; Moloney, R.D.; Clarke, G.; Dinan, T.G.; Cryan, J.F. The impact of microbiota on brain and behavior: Mechanisms & therapeutic potential. In Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 373–403.
- Averina O. V. et al. Bacterial metabolites of human gut microbiota correlating with depression //International journal of molecular sciences. – 2020. – p. 21. – №. 23. – p. 9234.
