
Гемофилия. История болезни от царских династий до наших дней.

С 1989 года 17 апреля установлен Всемирным днем гемофилии.
Инициатива проведения этого дня принадлежит сразу двум крупным медицинским организациям: Всемирной организации здравоохранения и Всемирной федерации гемофилии. Эта дата была выбрана не случайно, а является данью памяти основателю «Всемирной федерации гемофилии» – Франку Шнабелю и совпадает со днем его рождения.
Цель Всемирного дня гемофилии– повысить осведомленность населения Земли о гемофилии и других наследственных нарушениях свертываемости крови, а также улучшить доступность лечения и поддержку для людей, живущих с этими состояниями по всему миру.
В 2022 году более 257 тысячам человек во всем мире был установлен диагноз гемофилия. В то же время еще 100 тысяч человек живут с болезнью Виллебранда [1]. В России же, по данным НМИЦ гематологии Минздрава РФ насчитывается около девяти тысяч человек, страдающих гемофилией [2].
Печать королевских династий. История болезни.
О гемофилии человечеству известно уже более 2000 лет.
Первое литературное упоминание этого заболевания содержится в Вавилонском Талмуде: «Сообщалось о четырех сестрах из Сефориса. Первая произвела обрезание своему сыну, и он умер; вторая — и он умер; третья — и он умер. Тогда четвертая сестра пришла к рабби Симеону, сыну Гамалиеля, который сказал ей: откажись от обрезания, потому что есть семьи, у которых кровь жидкая, тогда как в других семьях она свертывается» [3].
В XII веке арабский врач Абу аль Касим первым в истории описал симптомы гемофилии. Он рассказал о семьях, в которых дети мужского пола умирали от небольших повреждений. А в 1793–1798 гг. немецкий врач Ров писал об аналогичных семействах в Вестфалии [3].
Первое же современное описание гемофилии появилось в 1803 году, когда американский врач Джон Конрад Отто описал наследственное нарушение свертываемости крови в нескольких семьях, где были больны только мужчины, которых он назвал «кровоточащими». А за несколько лет до этой публикации в журнале Салемский вестник (еженедельная газета, которая издевались в Салеме, штат Массачусетс, США) был описан случай 19-летнего мужчины, который умер от “обескровливания” так же, как и его пять братьев.
О наследственной природе заболевания в 1820 г. указывал Нассе в сочинении «О наследственной склонности к смертельным кровотечениям». Именно в этой статье был описан так называемый закон Нассе, который гласит: «гемофилия полностью передается не зараженными женщинами своим сыновьям»[6].
Первой известной носительницей «царской болезни» считается английская королева Виктория (1837–1901). Вероятнее всего, мутация гена произошла впервые в высокопоставленной семье именно в ее генотипе, так как до этого случаи гемофилии в династии зарегистрированы не были. Именно от Виктории это заболевание было получено царствующими семьями Германии, Испании и России. Гемофилией страдал один из сыновей Виктории (Леопольд, герцог Олбани), а также ряд других потомков. Пожалуй, самым известным случаем «царской болезни» стал царевич Алексей, сын российского царя Николая II. Такая связь с правящими семьями породила неофициальные названия гемофилии: «викторианская болезнь» и «царская болезнь».
Истинное литературное название болезни дал немецкий физиолог Феликс Хоппе в диссертации «О гемофилии, или наследственная склонность к смертельным кровотечениям», защищенной в 1928 г. в Вюрцбюрге. Термин «гемофилия» на самом деле означает «близость к крови», что в современных реалиях не совсем соответствует истине. Несмотря на упоминание Хоппе, немецкий врач Иоганн Лукас Шенляйн предпочитал использовать термин «геморрафилия» («склонность к кровотечению»), который более точно описывал симптоматику патологии. Однако впоследствии эти два термина стали использоваться как синонимы. Примечательно, что примерно в 1850-х годах термин «геморрафилия» вышел из употребления, и «гемофилия» стала предпочтительным обозначением
Несмотря на столь богатую историю, этиологию гемофилии установили лишь в 1861 году. Ученым, совершившим это открытие стал наш соотечественник Александр Александрович Шмидт. Он вывел ферментативную теорию семейной кровоточивости. Позже его предположения подтвердились: выяснилось, что в плазме крови больных не хватает некоторых белков-факторов свертываемости крови, которые есть у здоровых людей [3].
Первоначально считалось, что недостаточность свертывания крови, связанный с гемофилией, является результатом дефицита кальция. Впоследствии это было опровергнуто немецким терапевтом, физиологом Паулем Оскаром Моравицем(1908), предположившим, что этиологическим фактором является дефицит тромбокиназы. В 1911 году Томас Аддис, шотландский и американский врач, исследовал несколько факторов свертывания в крови и тканях, и пришел к выводу, что протромбин в крови больных гемофилией имеет дефекты в структуре. В более поздней публикации 1916 года он сообщил о сокращении времени свертывания крови больных гемофилией после внутривенного вливания свежей сыворотки крови[7-8].
Давние споры о том, является ли отсутствующим или дефицитным фактором при гемофилии протромбин или одно из его производных, разрешились с разработкой Армандом Квиком и его коллегами одноэтапного теста времени протромбина в 1935 году (ныне исследование носит имя своего создателя). Это позволило ученому и его команде продемонстрировать, что содержание протромбина в плазме больных гемофилией было нормальным, что впоследствии было подтверждено патологоанатомом Кеннетом Бринкхаусом, который в 1939 году обнаружил, что в крови больных гемофилией замедлена скорость превращения протромбина при нормальном его уровне.
Поскольку до появления эффективных методов лечения гемофилия часто ассоциировалась со смертельным заболеванием, сложился своеобразный стереотип о том, что в ее лечении обязательно проведение гемотрансфузий. Хотя переливание крови было впервые предложено Шенлейном в 1832 г., самая ранняя запись о его проведении этого метода, вероятно, принадлежит Сэмюэлю Армстронгу Лейну в 1840 году [9]. Лейн рассказал о прямом переливании 12 унций (примерно 350 мл) крови «полной молодой женщины» 11-летнему мальчику, у которого в течение 6 дней шла кровь после операции по поводу косоглазия. После переливания кровотечение остановилось, и мальчик выздоровел. В других публикациях часто упоминается переливание крови ягненка людям в Германии в конце IX века.
В качестве альтернативы переливанию крови известный хирург Эрнст фон Бергман предложил использовать модифицированный физиологический раствор, который был улучшен Сидни Рингером путем добавления электролитов. К сожалению, «раствор Рингера» обладал лишь ограниченной способностью поддерживать коллоидно-осмотическое давление, и, в свете открытий Эрнеста Генри Старлинга, в конечном итоге к нему были добавлены желатин и альбумин.
Разработка первого специального препарата «антигемофильного фактора» датирована 1911 годом, когда Томас Аддис приготовил особую фракцию крови путем подкисления плазмы. Позже, в 1916 году, он описал сокращение времени свертывания гемофильной крови после внутривенного введения свежей человеческой сыворотки [10]. В 1934 году британский патологоанатом Роберт Макфарлейн описал кровоостанавливающее действие яда гадюки Рассела при местном применении после хирургических процедур, удаления зубов, носовых кровотечений и раневых кровотечений у больных гемофилией, больных геморрагическим диатезом и у здоровых людей. Приготовленный яд стал коммерчески доступен под названием «Стипен» и иногда используется до сих пор.
В 1946 году Кон описал фракцию Кона I, которая, помимо высокого содержания фибриногена, обладала антигемофильной активностью. Однако ее эффективность была слишком низкой, чтобы стать полноценным лекарством для лечения гемофилии.
Внедрение в середине 1960-х годов концентратов фактора VIII, полученных из плазмы доноров, наконец, сделало возможной заместительную терапию факторами свертывания крови, что привело к резкому увеличению как качества жизни, так и продолжительности жизни людей с гемофилией А.
Взгляд современной медицины на гемофилию.
Гемофилия — это редкое нарушение свертываемости крови, вызванное отсутствием или дефектом функции белков – факторов свертывания крови. Существует два основных типа гемофилии:
- гемофилия А (дефицит фактора VIII);
- гемофилия В (дефицит фактора IX).
Гемофилия А встречается чаще, поражая примерно 1 из 5000 новорожденных мальчиков. Гемофилия В составляет 13% случаев заболевания.
Ранее выделяли гемофилию типа С (дефицит фактора XI). Ее уникальность в том, что она встречается в равной степени как у мальчиков, так и у девочек. В настоящее время этот вид болезни исключён из классификации, так как его клинические проявления значительно отличаются от А и В [4].
Мутации при гемофилии представлены в нескольких генах. Наиболее частая мутация — инверсия в гене F8, удаляющая карбоксильный конец фактора VIII. Другой интересный класс мутаций — обратная перестановка повторов L1 в гене. При гемофилии В происходят различные мутации гена F9. Для всех мутаций остаточная ферментативная активность комплекса факторов VIII и IX полностью соответствует тяжести клинических проявлений.
Гемофилия наследуется по Х-сцепленному рецессивному типу. У женщин-носительниц пораженного гена может наблюдаться легкое или бессимптомное течение из-за защитного эффекта второй Х-хромосомы. У некоторых из них симптомы впервые проявляются только во время каких-либо инвазивных процедур или родов.
Нормальный гемостаз наступает, когда уровни факторов VIII и IX составляют ≥ 50% от нормы. Если вспомнить функционирование нормальной системы гемостаза, станет понятно, что взаимодействие VIII и IX способствует активации X фактора. Который в свою очередь через каскад реакций способствует образованию сгустка фибрина.
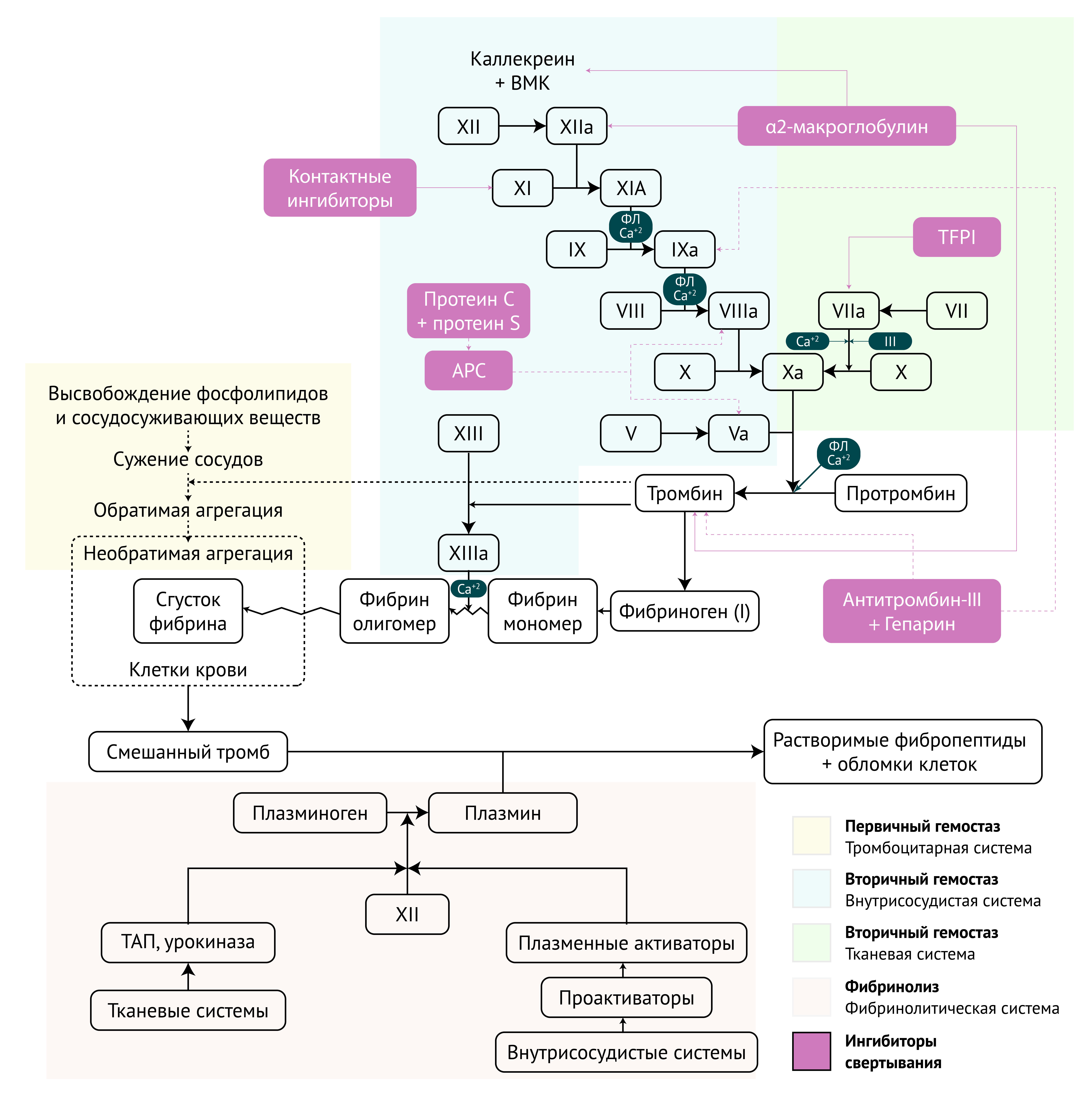
При отсутствии, недостатке или дисфункции факторов IX или VIII возникают нарушения гемостаза. Соответственно, при уровне факторов свертываемости крови 5%-40% устанавливается легкая степень течения гемофилии. От 1% до 5% - средняя. При уровне факторов меньше 1% гемофилия характеризуется тяжелым течением. Около 60–70% всех диагностированных случаев гемофилии составляют тяжелые формы заболевания [5].
Поскольку уровни фактора VIII также могут быть снижены при болезни Виллебранда, важно дифференцировать ее от гемофилии. У пациентов с впервые диагностированной гемофилией А в обязательном порядке пациентов измеряют активность фактора Виллебранда. Фактор Виллебранда - это молекулярный комплекс, обеспечивающий стабильность и защиту от преждевременной инактивации фактора VIII. При отсутствии фактора Виллебранда фактор VIII инактивируется раньше срока протеином C, и именно поэтому его уровень может быть значительно снижен.
В зависимости от степени тяжести гемофилия характеризуется различными симптомами.
Для тяжелого течения свойственно появление геморрагического синдрома уже на первом году жизни. Он характеризуется развитием гематом мягких тканей, кровоизлияниями в мышцы, посттравматическими кровотечениями из слизистых, часто гематурией. Серьезным симптомом тяжелого течения гемофилии является поражение, в основном, крупных суставов: коленных, голеностопных, локтевых, тазобедренных, с развитием гемартрозов. Могут развиваться жизнеугрожающие состояния, такие как кровоизлияния в мозг, забрюшинное пространство, плевральную и полость перикарда, в области шеи, а также желудочно-кишечные кровотечения [5].
Гемофилия средней тяжести имеет сходные проявления. Но первые признаки, как правило, развиваются после года, когда возрастает активность ребенка. При уровне факторов свертываемости крови больше 2% реже возникают кровоизлияния в суставы, забрюшинные гематомы, гематурии. Наиболее типичны посттравматические гематомы и длительные кровотечения, особенно при травмах слизистых оболочек, которые не останавливаются самостоятельно [5].
Легкая гемофилия может никак не проявляться на протяжении всей жизни. Геморрагический синдром обычно возникает вследствие значительных травм или при хирургическом лечении. Поражение опорно-двигательного аппарата встречается чрезвычайно редко [5].
Согласно клиническим рекомендациям РФ (2023) для установления диагноза «гемофилия»,необходимо наличие геморрагического синдрома в анамнезе у пациента и/или членов его семьи, а также 2 из 3 существующих критериев:
- отсутствие приобретенных коагулопатий;
- снижение активности FVIII/FIX ниже 50%;
- наличие мутаций генов FVIII или FIX.
К сожалению, гемофилия – наследственная неизлечимая болезнь. Но на данный момент, благодаря достижениям современной медицины, она успешно корректируется приемом заместительной терапии факторами свертывания крови. Современная терапия гемофилии базируется на принципе «домашнего лечения», обязательными условиями которого являются:
- наличие у пациента препаратов факторов свертывания крови (препарат находится там же, где пациент),
- решение о применении препарата принимает пациент или его родственники (если пациент несовершеннолетний) в соответствии с рекомендациями врача-гематолога,
- пациент и/или его родственники обучены правилам хранения и использования препаратов [5].
Обычно используют очищенные, вирус-инактивированные препараты, изготовленные из донорской плазмы человека, из группы «Факторы свертывания крови» (B02BD по АТХ классификации) – плазматические (содержащих или не содержащих фактор Виллебранда), рекомбинантные, или рекомбинантные концентраты факторов свертывания крови VIII/IX с пролонгированным периодом полувыведения. В настоящее время нет оснований для приоритетного выделения той или иной группы факторов свертывания крови. Но все же предпочтение должно отдаваться тому препарату, который при равной эффективности лучше всего переносится пациентом, имеет лучшие фармакокинетические индивидуальные показатели и наиболее удобен в использовании, исходя из конкретных объективных условий [5].
Список литературы
- Published by Statista Research Department, Nov 10, 2023
- Статья на сайте Министерства здравоохранения РФ. “Эксперт НМИЦ гематологии Минздрава России о современных возможностях лечения гемофилии” Зозуля Н.И. от 17 апреля 2022
- “Секреты наследственности человека” Афонькин Сергей Юрьевич
- “Царская болезнь: что необходимо знать о гемофилии” М.Качура, 2020. Журнал ZOOM
- Клинические рекомендации РФ 2023 (Россия). Наследственный дефицит фактора IX (D67), Наследственный дефицит фактора VIII (D66)
- Бринхаус К.М. Краткая история гемофилии с некоторыми комментариями к словуn«гемофилия». В: Бринкхаус К.М., Хемкер Х.К., редакторы. Справочник по гемофилии. Амстердам: Excerpta Medica 1975. с. 3–20.
- Аддис Т. Патогенез наследственной гемофилии. J Pathol Bacteriol 1911; 15: 427–
- Аддис Т. Влияние внутривенных инъекций свежей человеческой сыворотки и фосфатированной крови на время свертывания крови при наследственной гемофилии. Proc Soc Exp Biol Med 1916; 14: 19–23.
- Лейн С. Геморрагический диатез. Успешное переливание крови. Ланцет 1840;35:185–8.
- Аддис Т. Влияние внутривенных инъекций свежей человеческой сыворотки и фосфатированной крови на время свертывания крови при наследственной гемофилии.
