
Болезнь Ниманна-Пика

Ежегодно по всему миру появляются сообщения о миллионах новых случаев наследственных нейродегенеративных заболеваний, которые характеризуются прогрессирующим развитием дисфункции нервной системы, неизбежно приводящей к инвалидизации пациента и раннему летальному исходу. Системные наследственные болезни лизосомального накопления, протекающие с дегенеративными изменениями в центральной нервной системе, в частности - болезнь Ниманна-Пика - являются областью особого интереса среди исследователей- неврологов.
Болезнь Ниманна-Пика – гетерогенная группа прогрессирующих нейровисцеральных лизосомных липидных болезней накопления, характеризующаяся отложением сфингомиелина и других липидов преимущественно в ретикулоэндотелиальной и нервной ткани. В 1958 г. Crocker и Farber показали вариабельность степени выраженности клинических проявлений, возраста начала заболевания и интенсивности накопления липидов в тканях. Данное наблюдение привело к выделению четырех классификационных групп A-D в составе болезни Ниманна-Пика.
Тип А характеризуется ранним тяжелым поражением ЦНС и массивным повреждением тканей в результате патологического клеточного накопления.
Тип В, напротив, поражает преимущественно паренхиматозные органы. Для типов С и D (выделен из группы С специально для описания проявлений болезни в группе больных, проживающих на территории Новой Шотландии в Канаде) характерны умеренность висцеральных проявлений, подострое поражением нервной системы и относительно медленное прогрессирование, в сравнении с предыдущими типами.
О типах А и Б известно немного. Наиболее часто данные группы болезни Ниманна-Пика встречаются у евреев Ашкенази. Обнаружено, что в основе данных типов болезни лежат мутации в гене SMPD1, кодирующего фермент кислую сфингомиелиназу. Почти полное отсутствие конкретных данных, касающихся этиологии и патогенеза этих типов заболевания, связано с их низкой распространенностью и выраженной тяжестью симптомов, приводящих к смерти больных в первые месяцы после рождения.
Болезнь Ниманна-Пика, тип С (БНП-С) – наиболее распространенный тип заболевания. Наследуется по аутосомно-рецессивному типу и встречается с частотой 1:120 000 случаев живых новорожденных. Достоверной связи с этническим происхождением не имеет. Время начала манифестации заболевания, а также продолжительность жизни значительно варьируются даже у сибсов. Большая часть летальных исходов приходится на промежуток между 10 и 25 годами. С момента проявления первых неврологических симптомов до консультации со специалистом и постановки диагноза проходит в среднем 5-6 лет, что связано с низкой специфичностью и изолированностью начальных признаков. Значительная отсрочка в диагностике БНП-С, естественно, осложняет последующий процесс коррекции состояния.
Этиология
За возникновение БНП-С ответственны два гена- NPC1 и NРС2. Приблизительно в 95% случаев у пациентов наблюдается мутация в NPC1. Данный ген расположен на хромосоме 18 q11-q12 и состоит из 25 экзонов; его полная последовательность составляет 57052 kb NPC1 кодирует мембранный 13-ти доменный гликопротеин, состоящий из 1278 аминокислот и расположенный преимущественно в составе оболочки поздних эндосом. Основная функциональная стерол-чувствительная последовательность (615-797 аминокислоты) гомологична другим белкам, участвующим в липидном обмене- HMG-CoA редуктазы и SREBP (sterol regulatory element-binding protein). Кроме того, в структуре белка-продукта гена NPC1 имеются две интралюминальные последовательности, отвечающие за белок-белковые взаимодействия. Первая из них (аминокислотная последовательность 855-1098) представляет собой цистеин-содержащую петлю с мотивом типа "цинковый палец" и является мишенью 1/3 всех мутаций в исходном гене; вторая - высококонсервативный N-концевой домен, содержащий мотив "лейциновой застежки-молнии" (последовательность 25-264). К настоящему времени имеются сообщения о приблизительно 60 полиморфизмах и 334 мутациях в данном гене (данные Human Gene Mutation), из них 228 - миссенс- и нонсенс-мутации, 46 малых и 7 больших делеций, 26 мутаций, влияющих на сплайсинг, 24 малые и 1 большая инсерции. В этот перечень входят так же мутации в интронных последовательностях, которые создают дополнительные сайты сплайсинга. В литературе, посвященной генотип-фенотипической мутационной зависимости, имеются данные об отмеченной корреляции между тяжестью заболевания и нонсенс-мутациями, а также изменениями, связанными со сдвигом рамки считывания. Миссенс-мутации, в свою очередь, как правило, ответственны за нарушения функционирования стерол-связывающего домена и цистеиновой петли (клинически для этого типа нарушений характерно наиболее легкое течение заболевания).
Мажорных мутаций для данного гена нет, однако среди лиц западноевропейского происхождения самыми частыми обнаруживаемыми мутациями являются p.I1061T и p.P1007A, которые определяются в 20-25% случаев и в гомозиготном состоянии предрасполагают к развитию классической ювенильной формы БНП-С. В гетерозиготном состоянии мутация способствует проявлению симптомов болезни в более позднем возрасте. В оставшихся 5% случаев причиной заболевания служит изменение NPC2 (хромосома 14 q24.3, 5 экзонов) и его продукта - растворимого лизосомального белка, состоящего из 132 аминокислот и имеющего стерол-чувствительную гидрофобную высокоафинную (Kd = 30–50 nM) последовательность ("карман") для связывания холестерола в стехиометрическом соотношении 1:1.
Некоторыми авторами отмечена вероятная способность взаимодействия продуктов генов обоих типов между собой в процессе выполнения ими их функции в метаболизме холестерола (а именно - транспорте его из лизосом). Известно, что в нормальных клетках липопротеины низкой плотности, поглощенные путем эндоцитоза ЛПНП, поступают в поздние эндосомы/лизосомы, где гидролизуются с высвобождением свободного холестерола, который, в свою очередь, быстро транспортируется из эндосом к мембране.При БНП-С поломка любого из генов NPC приводит к специфическому разобщению процессов, участвующих в метаболизме и утилизации захваченного путем эндоцитоза холестерола, что приводит к его накоплению в лизосомах клеток паренхиматозных органов и к вторичному нарушению обмена сфинголипидов (в частности - гликосфинголипидов) в нервной ткани. Последнее наблюдение заслуживает отдельного пристального внимания, так как адекватное функционирование систем, поддерживающих метаболизм холестерола, имеет большое значение в нормальной жизнедеятельности нейронов центральной нервной системы (головной мозг человека содержит 25% всего неэстерифицированного холестерола, находящегося в организме).
При БНП-С в ЦНС происходит нарушение эстерификации холестерола и отложение как его, так и ганглиозидов GM2, GM3, а также бис-моноацилглицеролфосфата в поздних эндосомах/лизосомах. Кроме того, происходит увеличение размеров ранних эндосом и повышение уровня лизосомальных гидролаз,в частности катепсина D.Особенно ярко эти процессы выражены в ЦНС, в микроглии, нейронах ствола мозга и клетках Пуркинье мозжечка. Тем не менее, несмотря на активное накопление холестерола в нейронах и глие, общее содержание холестерола в нервной ткани значительно не повышается. Вероятно, это происходит потому, что при БНП-С интенсивность синтеза холестерола значительно снижается, за счет замедления процессов образования ключевых ферментов.
С процессом накопления холестерола в ЦНС так же тесно связан феномен усиления аутофагии. Данная зависимость была доказана в результате обнаружения иммунопозитивных скоплений белка- маркера аутофагии -LC3 в непосредственной близости от филипин-окрашеного холестерола. Было отмечено увеличение числа аутофагосом в клетках, а так же развитие нейровоспалительных процессов, приводящих к нейродегенерации. Тем не менее, к настоящему моменту не существует четкого объяснения взаимосвязи всех наблюдаемых изменений в метаболизме холестерола и, несмотря на очевидность участия поломки в генах NPC в развитии дисфункции, конкретная роль белков-продуктов этих генов в описанных патологических событиях еще не определена.
Клиническая картина
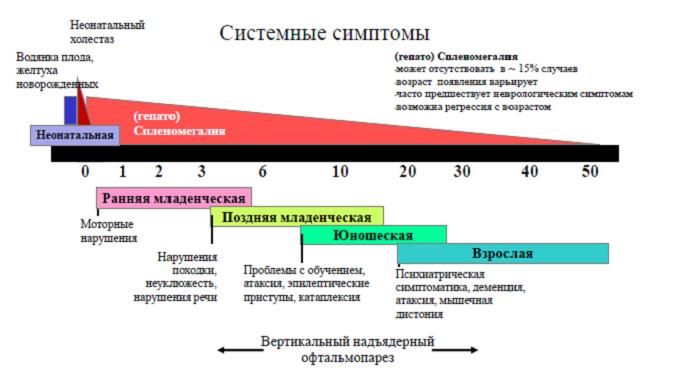 Клинические проявления болезни Ниманна-Пика, тип С [Vanier M. «Niemann–Pick disease type C» , Clinical genetics, 2003]
Клинические проявления болезни Ниманна-Пика, тип С [Vanier M. «Niemann–Pick disease type C» , Clinical genetics, 2003]
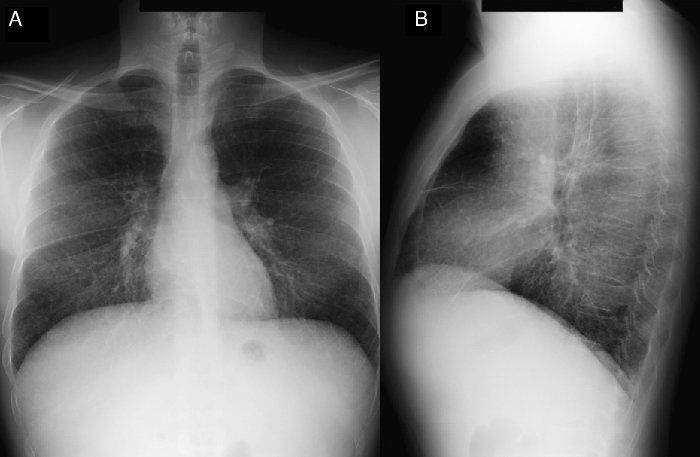
Заболевание носит системный характер и преимущественно поражает селезенку, печень и головной мозг, что выражается в ряде висцеральных, неврологических и психиатрических нарушений. Именно своеобразное сочетание проявлений дисфункции этих органов позволяет заподозрить наличие у пациента Болезни Ниманна-Пика, тип С в процессе дифференциального диагноза. Висцеральные проявления включают в себя спленомегалию, гепатомегалию, длительную холестатическую желтуху новорожденных, фетальную водянку и асцит, а также спектр легочных патологий (альвеолярный липидоз, интерстициальная пневмония).
Начало заболевания, особенно в случае манифестации в раннем возрасте, чаще всего сопряжено с изолированным или сочетанным с гепатомегалией увеличением селезенки, которое, поначалу, до проявления первых неврологических симптомов, не находит рационального объяснения со стороны клиницистов. Спленомегалия при болезни Ниманна-Пика носит продолжительный характер. Выраженность увеличения варьирует от едва заметного до значительного и, что примечательно, не находится в корреляции со степенью тяжести сопутствующего неврологического дефицита и стадией заболевания. Кроме того, изолированное отсутствие спленомегалии не является показанием для исключения БНП-С.
Изолированное увеличение печени без вовлечения селезенки в большинстве случаев характерно для позднего начала заболевания и часто обнаруживается только в процессе ультразвукового исследования. В целях постановки диагноза необходимо проведение дифференциальной диагностики с наследственными обменными болезнями - мукополисахаридозами, гликогенозами и другими типами болезни Ниманна-Пика.
Длительная холестатическая желтуха новорожденных может служить индикатором рано проявляющейся БНП-С. Тяжесть ее может варьироваться от транзиторной гипербилирубинемии (значение конъюгированного билирубина > 1.2 мг/дл) до значительного застойного повреждения печени с высокой вероятностью летального исхода в течение первого года жизни. Имеются данные о спонтанной регрессии явлений холестаза ко 2-4 месяцам жизни, однако изменения со стороны печени персистируют в течении более длительного периода времени. При подозрении на БНП-С по наличию у ребенка продолжительной холестатической симптоматики, в особенности в случае ее сочетания со спленомегалией, необходимо исключение идиопатического неонатального гепатита и биллиарной атрезии.
Водянка плода как проявление БНП-С встречается нечасто и имеет неимунную природу. Данный признак обнаруживается при ультразвуковом исследовании в антенатальном периоде в виде генерализованного отека с преимущественным скоплением жидкости в перикардиальном пространстве и брюшной полости (асцит). Вследствие низкой специфичности, обнаружение выпотных явлений в процессе диагностики требует рассмотрения вероятности проявления не только генетической патологии (в том числе и болезни Ниманна-Пика), но и инфекционного поражения, гемоглобинопатии и пороков сердца. Поражение легких, как и тромбоцитопения, встречается при тяжелом течении заболевания и дополняет проявления, связанные с выраженной гепато- и спленомегалией, создавая дополнительные предпосылки для подозрения на БНП-С. (рис.1)
Неврологически структуру заболевания составляют вертикальный надъядерный паралич взора, церебеллярная атаксия и геластическая катаплексия. Другие неврологические нарушения- дизартрия, дистония, дисфагия, эпилептические припадки и нейросенсорная тугоухость- имеют более низкую специфичность и в дебюте заболевания встречаются реже. Вертикальный надъядерный паралич взора представляет собой патологическое изменение саккадических движений глаз в вертикальной плоскости и является наиболее характерным признаком БНП-С. Причиной нарушения саккад в данном случае является повреждение нейрональных структур в ростральных интерстициальных ядрах медиального продольного пучка, а позднее и вестибулярных ядер, находящихся под влиянием мозжечка. Клинически этот симптом проявляется в неспособности обращения взора вверх/вниз без изменения положения головы, что значительно нарушает качество жизни пациента. Причиной атаксии является поражение клеток Пуркинье мозжечка. При этом пациенты испытывают трудности в ходьбе, неуклюжи и склонны к частым падениям. Как и в случае других заболеваний, проявляющихся атаксией (с которыми обязательно проведение дифференциального диагноза), больные оказываются неспособны к выполнению мозжечковых тестов при осмотре, что в сочетании с вертикальным параличом взора и/или имеющейся висцеральной симптоматикой позволяет заподозрить БНП-С.
Кратковременные эпизоды резкой потери мышечного тонуса, не сопровождающиеся утратой сознания – геластическая катаплексия- появляются, в среднем, с двухлетнего возраста и провоцируются эмоциональной активностью (во время смеха, плача, при испуге). Важной диагностической особенностью состояния является отсутствие ЭЭГ нарушений, которые могли бы быть характерны для эпилептических припадков.
По результатам практических наблюдений, было показано, что клиническая картина и прогноз находятся в зависимости от возраста начала заболевания. В связи с этим, по характерным для БНП-С проявлениям (в том числе и неврологическим) в структуре болезни выделяют несколько форм: перинатальная (до 2 месяцев), ранняя младенческая (от 2 месяцев до 2 лет), поздняя младенческая (от 2 до 6 лет), ювенильная (6-12/15 лет), взрослая (старше 12/15 лет).
Для перинатальной формы характерно превалирование описанной выше висцеральной симптоматики над неврологической, возможно отчасти потому, что дисфункция нервной системы, проявляющаяся в данном периоде носит тяжелый характер и приводит к смерти еще до подозрения на диагноз БНП-С.
Ранняя младенческая форма характеризуется развитием гепатоспленомегалии, проявлением признаков центральной гипотонии в качестве дебютного симптома, а также задержкой моторного развития, тяжесть которой стремительно увеличивается ко второму году жизни и сопровождается нарастающей (по мере вовлечения в патологический процесс пирамидного тракта) спастичностью. Дисфункция двигательной системы сопровождается явлениями интенционного тремора, что так же затрудняет освоение моторных задач. Многие пациенты с младенческой формой БНП-С навсегда утрачивают возможность самостоятельного передвижения. Кроме того, отмечено и замедление интеллектуального развития.
При начале манифестации заболевания в возрастном периоде от 2 до 6 лет отмечаются проявления атаксии: нарушение походки, склонность к частым падениям. По мере нарушения функции нервной системы появляются дизартрия, дисфагия (вплоть до необходимости проведения гастростомии), мышечная спастичность; прогрессирует когнитивный дефицит. Отмечается задержка развития речи и нарушение слуха (нейросенсорная тугоухость). Впервые в этом возрасте возникают эпизоды геластической катаплексии; часты генерализованные и парциальные эпилептические припадки. Присутствие последних в клинической картине является признаком неблагоприятного прогноза для жизни и высокой вероятности летального исхода к возрасту 7-12 лет.
Ювенильная форма в литературе признана классической. Первые признаки заболевания связаны с увеличением селезенки.Этот симптом, однако, получает должную оценку, как правило, только при ретроспективном анализе. Маркерными же являются неврологические проявления - сочетания вертикального надъядерного паралича взора, катаплексии, атаксических признаков, дизартрии и дисфагии. Обращают на себя внимание прогрессирующий когнитивный дефицит и психические расстройства. Продолжительность жизни зависит от скорости нарастания дисфункции нервной системы и выраженности висцеральных проявлений, но обычно не превышает 30 лет.
В случае манифестации БНП-С во взрослом возрасте диагноз ставится, как правило, на основании наличия необъяснимой другими причинами гепатомегалии в сочетании с положительными диагностическими, в т.ч. генетическими, тестами на маркеры болезни. Для позднего начала в структуре заболевания характерно превалирование психических нарушений– психозов, депрессий, эпизодов агрессивного поведения, параноий, слуховых и зрительных галлюцинаций, навязчивых состояний. Клинически значимого увеличения селезенки, как правило, не наблюдается. Неврологические симптомы ограничиваются надъядерным параличом взора и атактическими расстройствами движения.
Диагностика
Заподозрить болезнь Ниманна-Пика, тип С возможно при обнаружении комплекса висцеральных, неврологических и психиатрических симптомов во время клинического исследования. Необходимо проведение неврологической оценки мышечного тонуса и силы, проверки двигательных рефлексов, выявление особенностей походки, определение степени когнитивных и психических нарушений при тестировании. В процессе оценки функции органа зрения особенное значение придается выявлению дефицита взора в вертикальной плоскости.
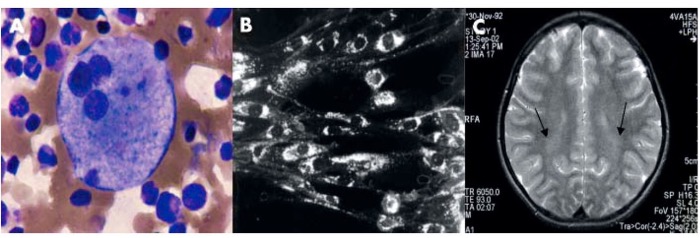
Целесообразно определение остроты слуха, проведение анализа аудиограмм и исследование вызванных слуховых потенциалов. Выявленные при наблюдении характерные симптомы анализируются в сумме с учетом шкалы индекса вероятности БНП-С, включающей в себя оценку всех неврологических, висцеральных и психиатрических нарушений по баллам. Кроме того, производится учет данных семейного анамнеза и признаков, указывающих на наследственный характер заболевания. Если сумма баллов составляет 70 и более, следует считать вероятным диагноз болезнь Ниманна-Пика, тип С, а также необходимо в срочном порядке направить пациента на прохождение медико-генетического консультирования и молекулярно-генетического анализа. При сумме баллов менее 40 подозрение на БНП-С считается необоснованным. Инструментальное МРТ и КТ исследование позволяет определить мозжечковую и корковую атрофию или, в случае тяжелых ранних младенческих форм, и дегенеративные изменения в белом веществе. (рис.2С) Часто в среднем саггитальном изображении отмечается уменьшение объема среднего мозга, а так же атрофия мозолистого тела.
Необязательным, но способствующим корректной диагностике исследованием является морфологическая оценка аспирационных биоптатов костного мозга, кожи и печени. (рис. 2А). При рассмотрении препаратов костного мозга обнаруживается множество "голубоватых" гистиоцитов, а также тучных клеток, перегруженных липидными отложениями и селективно положительно окрашивающихся на белок филипин; в случае исследовании печеночных биоптатов выявляют признаки холестатического поражения, множество тучных портальных макрофагов, купферовских клеток, отложения гранул липофусцина.
При наличии у пациента признаков холестаза или гиперспленизма имеет смысл проведение биохимических анализов, которые, помимо небольшого неспецифического повышения трансаминаз, выявляют пониженную концентрацию ЛПВП, повышенное содержание триглицеридов, а также характерное повышение активности сывороточной хитотриозидазы и кислой сфингомиелиназы (в лейкоцитах). Кроме того, в настоящее время для первичного скрининга широко применяется исследование уровня оксистеролов (D7-3,5,6-triol и 3β, 5α, 6β-Trihydrooxycholestane-d7) в плазме крови.
Нарушение транспорта холестерола и липидного равновесия может быть выявлено и продемонстрировано при использовании филипинового теста, проводимого на культуре фибробластов кожи в немногочисленных специализированных центрах. Фибробласты культивируются в среде, обогащенной ЛПНП, а затем окрашиваются на филипин (флуоресцентное вещество, полученное из Streptomyces filipinensis и образующее специфические комплексы совместно с неэстерифицированным холестеролом). Как правило (80-85% случаев), флуоресцентное микроскопическое исследование NPC-положительных клеток обнаруживает большое количество флуоресциирующих перинуклеарных везикул, наполненных неэстерифицированным холестеролом. (рис. 2В). Менее выраженное окрашивание описано у гетерозиготных носителей и у пациентов с вариантным биохимическим фенотипом -результатом ассоциации нескольких рекуррентных мутаций NPC, а также встречается среди больных, страдающих от недостаточности сфингомиелиназы, обусловленной другими причинами. Важно отметить, что выраженность филипиновой реакции не коррелирует со степенью тяжести клинических проявлений БНП-С.
Болезнь Ниманна –Пика, тип С является наследственным аутосомно-рецессивным заболеванием, поэтому для подтверждения диагноза настоятельно рекомендуется проведение генетического анализа во всех случаях подозрения на данную патологию. В основе генетического анализа при БНП-С лежит прямое секвенирование экзонов NPC1 (25 экзонов), реже NPC2 (5 экзонов), вследствие наиболее частой встречаемости локализации мутации в первом упомянутом гене. Мажорных мутаций для данного заболевания не существует, однако отмечена высокая частота встречаемости генетических изменений в 12-22 экзонах гена NPC1.
При возможности, должно проводиться молекулярно- генетическое исследование ближайших родственников для того, чтобы удостовериться в расщеплении аллелей и подтверждении наличия гомозиготного статуса.
Лечение
Специфического лечения для болезни Ниманна-Пика типов А и В не существует. Имеются исследовательские данные об относительной эффективности пересадки донорских органов взамен утративших свою функцию в результате избыточного липидного накопления с целью продления и улучшения качества жизни у отдельных пациентов. Тем не менее, по понятным причинам, данный способ «лечения» не может применен повсеместно и дальнейшие перспективы в терапии связывают исключительно с разработкой генной терапии. Болезнь Ниманна-Пика, тип С- прогрессирующее заболевание с очень высокой вероятностью ранней детской смерти или скорого летального исхода в случае ювенильных и взрослых форм (за исключением всего 3х задокументированных случаев, когда БНП-С манифестировала в возрасте старше 53 и не сопровождалась увеличением селезенки). Тем не менее, скорость прогрессирования отдельных групп симптомов и качество жизни являются терапевтически частично корригируемыми факторами.
Основное лечение при БНП-С – симптоматическое. Единственное доступное в настоящее время специфическое лечение осуществляется ингибитором гликозилцерамид-синтазы – препаратом Miglustat («Zavesca», Actelion Pharmaceuticals Ltd.). Препарат способен проникать через гематоэнцефилический барьер. При приеме Миглустата отмечено замедление прогрессирования неврологической симптоматики и увеличение продолжительности жизни на 20% в случае начала лечения на ранних стадиях заболевания. Однако, препарат не оказывает значительного влияния на выраженность системных висцеральных проявлений. Таким образом, именно прогрессирование органной недостаточности (легочный фиброз, дыхательная недостаточность и поражение печени) становится основной причиной смерти пациентов. С 2009 года в FDA рассматривается проект по изучению эффективности 2-гидроксипропил-β-циклодекстрина, способного связывать холестерин, уменьшать выраженность накопительного процесса в печени и селезенке и замедлять процесс нейродегенерации при болезни Ниманна-Пика, однако план клинического исследования до сих пор находится на стадии разработки.
В некоторых исследованиях предполагается благоприятное влияние куркумина на внутриклеточный гомеостаз кальция и метаболизм липидов. Так, повышение концентрации кальция в цитоплазме под действием куркумина вызывало нормализацию клеточного фенотипа и увеличение выживаемости у мышей с мутацией в гене NPC1.
Таким образом, Болезнь Ниманна-Пика, тип С – малоизученное наследственное заболевание с недостаточно известным механизмом нарушения липидного обмена.Сравнительно низкая частота встречаемости патологии, а так же отсутствие характерных исключительно для данного заболевания клинических признаков приводит к возникновению значительной отсрочки в диагностике БНП-С, что осложняет последующий процесс коррекции состояния и отрицательно сказывается на качестве и продолжительности жизни пациента. Большая часть рекомендованных общеклинических диагностических исследований не предоставляют достоверных данных, свидетельствующих в пользу диагноза болезнь Ниманна-Пика, тип С, не позволяют судить о клиническом типе и планировать терапию.Перспективы в исследовании закономерностей развития данной патологии связаны с разработкой методов биохимической диагностики и молекулярно-генетического анализа и подходов к лечению ( в том числе - генной терапии) с целью максимально возможного улучшения качества и продолжительности жизни пациентов.
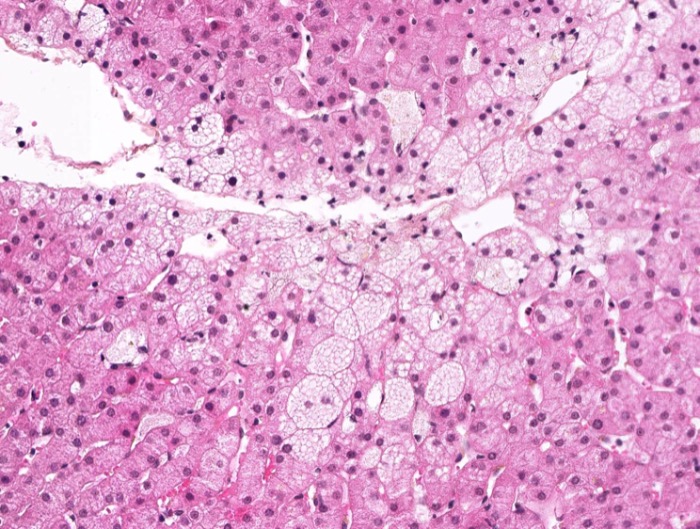
Болезнь Ниманна-Пика,тип В. Тучные клетки в препарате селезенки.
Литература:
- Захарова Е. Ю. и др. Клинико-генетические особенности болезни Ниманна-Пика, тип С //Вестник Российской академии медицинских наук. – 2012. – №. 12. – С. 60 - 65.
- Клюшников С.А. Алгоритмы диагностики болезни Ниманна-Пика, тип С //Нервные болезни. - 2012. - №3. – С. 8-12.
- Клюшников С. А. Болезнь Ниманна-Пика, тип С - лизосомная патология с нарушением внутриклеточного транспорта липидов //Нервные болезни. – 2014. – №. 1. – С. 4-14.
- Михайлова С. В. и др. Болезнь Ниманна-Пика тип С. Клинические примеры //Педиатрическая фармакология. – 2010. – Т. 7, №. 5. – С. 8-17.
- Михайлова С. В., Захарова Е. Ю., Петрухин А. С. Нейрометаболические заболевания у детей и подростков //Диагностика и подходы к лечению. - М.: Литтерра, 2011. – 289с.
- Abel L. A. et al. Saccades in adult Niemann-Pick disease type C reflect frontal, brainstem, and biochemical deficits //Neurology. – 2009. – Vol. 72, №. 12. – P. 1083-1086.
- Archer T. et al. Staging neurodegenerative disorders: structural, regional, biomarker, and functional progressions //Neurotoxicity research. – 2011. – Vol. 19,№. 2. – P. 211-234.
- Aqul A. et al. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment //The Journal of Neuroscience. – 2011. – Vol. 31, №. 25. – P. 9404-9413.
- Bauer P. et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study //Human molecular genetics. – 2013.- Vol.2, №. 1 – P.284.
- Bi X., Liao G. Cholesterol in Niemann-Pick Type C disease // Subcell Biochem. – 2010. – Vol.51. – P. 319-335.
- Bergamin N. et al. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient’s skin //Orphanet J Rare Dis. – 2013. – Vol. 8, №. 1. – P. 34-34.
- Bonnot O. Niemann-Pick Disease Type C: an Example of an Inborn Error of Metabolism Producing Psychiatric Manifestations //European psychiatric rewiew. – 2011. - Vol.4, №.3.- P.84-88.
- Carstea E. et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis //Science. – 1997. – Vоl. 277, №. 5323. – P. 228-231.
- Chien Y. et al. Long-term efficacy of miglustat in paediatric patients with Niemann-Pick disease type C //Journal of inherited metabolic disease. – 2013. – Vol. 36, №. 1. – P. 129-137.
- Church H. et al. Abnormal filipin staining and phenotypic overlap between acid sphingomyelinase deficiency and Niemann-Pick C disease //Molecular Genetics and Metabolism. – 2014. – Vol. 2, №. 111. – P. 32.
- Elrick M. J. et al. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease //Human molecular genetics. – 2012. – Vol. 36, №. 9. – P. 324.
- Fan M. et al. Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling //Journal of lipid research. – 2013. – Vol. 54, №. 10. – P. 2800-2814.
- Fancello T. et al. Molecular analysis of NPC1 and NPC2 gene in 34 Niemann–Pick C Italian patients: identification and structural modeling of novel mutations //Neurogenetics. – 2009. – Vol. 10, №. 3. – P. 229-239.
- Gabande-Rodriguez E. et al. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A //Cell Death & Differentiation. – 2014. – Vol. 21,№. 6. – P. 864-875.
- Garver W. et al. The National Niemann–Pick C1 disease database: report of clinical features and health problems //American journal of medical genetics Part A. – 2007. – Vol. 143, №. 11. – P. 1204-1211.
- Griese M. et al. Respiratory disease in Niemann‐Pick type C2 is caused by pulmonary alveolar proteinosis //Clinical genetics. – 2010. – Vol. 77, №. 2. – P. 119-130.
- Infante R. et al. Purified NPC1 protein I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein //Journal of Biological Chemistry. – 2008. – Vol. 283, №. 2. – P. 1052—1063.
- Ishibashi S., Yamazaki T., Okamoto K. Association of autophagy with cholesterol-accumulated compartments in Niemann-Pick disease type C cells //Journal of Clinical Neuroscience. – 2009. – Vol. 16, №. 7. – P. 954-959.
- Iturriaga C. et al. Niemann–Pick C disease in Spain: clinical spectrum and development of a disability scale //Journal of the neurological sciences. – 2006. – Vol. 249, №. 1. – P. 1-6.
- Kelly D. et al. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease //The Journal of pediatrics. – 1993. – Vol. 123, №. 2. – P. 242-247.
- King K. et al. Hearing Loss is an Early Consequence of Npc1 Gene Deletion in the Mouse Model of Niemann–Pick Disease, Type C //Journal of the Association for Research in Otolaryngology. – 2014. – Vol. 15, №. 4. – P. 529-541.
- Lopez M. Genetic dissection of a cell-autonomous neurodegenerative disorder: lessons learned from mouse models of Niemann-Pick disease type //Dis. Model. Mech. - 2013. - Vol.6. - P. 1089-1100.
- Lorenzoni P. et al. Niemann-Pick disease type C: a case series of Brazilian patients //Arquivos de neuro-psiquiatria. – 2014. – Vol. 72, №. 3. – P. 214-218.
- Ludolph A. et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options //European Journal of Neurology. – 2009. – Vol. 16,№. 3. – P. 297-309.
- Macías Vidal J. et al. Molecular analysis of 30 Niemann–Pick type C patients from Spain //Clinical genetics. – 2011. – Vol. 80, №. 1. – P. 39-49.
- McKay Bounford K., Gissen P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C // J. Neurol. – 2014. – Vol. 216. – P. 569- 575.
- Mengel E. et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description // Orphanet J Rare Dis. – 2013. – Vol. 8. – P. 166.
- Pedroso J. et al. Teaching video neuroimages: gelastic cataplexy as the first neurologic manifestation of Niemann-Pick disease type C //Neurology. – 2012. – Vol. 79, №. 22. – P. 189-189.
- Patterson M. et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry //Orphanet J Rare Dis. – 2013. – Vol. 8, №. 12. –P. 10.1186.
- Porter F. et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease //Science translational medicine. – 2010. – Vol. 2, №. 56. – P.681.
- Ramirez C. et al. Ontogenic changes in lung cholesterol metabolism, lipid content, and histology in mice with Niemann–Pick type C disease //Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. – 2014. – Vol. 1841, №. 1. – P. 54-61.
- Rosenbaum A. , Maxfield F.. Niemann‐Pick type C disease: molecular mechanisms and potential therapeutic approaches //Journal of neurochemistry. – 2011. – Vol. 116, №. 5. – P. 789-795.
- Salsano E. et al. Vertical supranuclear gaze palsy in Niemann-Pick type C disease //Neurological Sciences. – 2012. – Vol. 33.№. 6. – P. 1225-1232.
- Sarna J. et al. Patterned Purkinje cell degeneration in mouse models of Niemann‐Pick type C disease //Journal of Comparative Neurology. – 2003. – Vol. 456, №. 3. – P. 279-291.
- Sévin M. et al. The adult form of Niemann–Pick disease type C //Brain. – 2007. – Vol. 130, №. 1. – P. 120-133.
- Solomon D. et al. Niemann‐Pick Type C Disease in Two Affected Sisters: Ocular Motor Recordings and Brain‐Stem Neuropathology //Annals of the New York Academy of Sciences. – 2006. – Vol. 1039, №. 1. – P. 436-445.
- Stampfer M. et al. Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators //Orphanet J Rare Dis. – 2013. – Vol. 8, №. 1. – P. 35.
- Staretz-Chacham O. et al. Lysosomal storage disorders in the newborn //Pediatrics. – 2009. – Vol. 123, №. 4. – P. 1191-1207.
- Stein V. et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C //Journal of neuropathology and experimental neurology. – 2012. – Vol. 71, №. 5. – P. 434.
- Vance J. Lipid imbalance in the neurological disorder, Niemann-Pick C disease //FEBS letters. – 2006. – Vol. 580, №. 23. – P. 5518-5524.
- Vanier M., Millat G. Niemann–Pick disease type C //Clinical genetics. – 2003. – Vol. 64, №. 4. – P. 269-281.
- Vanier M. Complex lipid trafficking in Niemann-Pick disease type C //Journal of inherited metabolic disease. – 2015. – Vol. 38,№. 1. – P. 187-199.
- Walterfang M. et al. Dysphagia as a risk factor for mortality in Niemann-Pick disease type C: systematic literature review and evidence from studies with miglustat //Orphanet J Rare Dis. – 2012. – Vol. 7, №. 1. – P. 76.
- Wraith J. et al. Niemann-Pick type C Suspicion Index tool: analyses by age and association of manifestations //Journal of inherited metabolic disease. – 2014. – Vol. 37,№. 1. – P. 93-101.
- Wraith J. et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C //Molecular genetics and metabolism. – 2009. – Vol. 98, №. 1. – P. 152-165.
- Wraith J. Imrie J. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat //Therapeutics and clinical risk management. – 2009. – Vol. 5. – P. 877.
- Yanjanin N. et al. Linear clinical progression, independent of age of onset, in Niemann–Pick disease, type C //American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. – 2010. – Vol. 153,№. 1. – P. 132-140.
- Zervas M., Dobrenis K., Walkley S. Neurons in Niemann‐Pick Disease Type C Accumulate Gangliosides as Well as Unesterified Cholesterol and Undergo Dendritic and Axonal Alterations //Journal of Neuropathology & Experimental Neurology. – 2007. – Vol. 60, №. 1. – P. 49-64.
- Zhang M. et al. Differential trafficking of the Niemann‐Pick C1 and 2 proteins highlights distinct roles in late endocytic lipid trafficking //Acta paediatrica. – 2008. – Vol. 92, №. 443. – P. 63-73.
