
Гемолитико-уремический синдром
В 1955 г. C. Gasser с соавторами описали наблюдавшееся ими у 5 детей заболевание, которое представляло собой сочетание острой почечной недостаточности (ОПН) с гемолитической анемией и тромбоцитопенией, развивающимися на фоне инфекционной диареи (E. coli, Sh. dysenteriae, S. pneumoniae), и назвали его гемолитико-уремическим синдромом (ГУС).
Эпидемиология
С данным синдромом должен быть знаком каждый педиатр, ведь ГУС является основной причиной развития ОПН у детей до 3-х лет. Частота возникновения ГУС у детей данного возраста составляет 2–3 случая на 10 тысяч детей. Причем в разных регионах заболеваемость типичным ГУС (тГУС) значительно варьирует (в зависимости от численности сельского населения, особенностей водоснабжения — в Аргентине и Уругвае эшерихиоз эндемичен, поэтому частота достигает 10 случаев на 100 тыс. населения в год; в более холодных регионах заболеваемость также выше — в Шотландии, по сравнению с Англией, в 2 раза выше — 3,4 vs 1,54 на 100 тыс. населения в год). Для тГУС чаще характерны эпидемические подъемы заболеваемости, но могут быть и спорадические случаи (более характерно для атипичного ГУС). Резервуаром инфекции являются фекалии крупного рогатого скота (E. coli O157 длительно выделяют в стуле Shiga like toxin (Stx) 2 типа). Человек заражается при употреблении сырой телятины, непастеризованного молока, загрязненных фруктов и овощей, контаминированной воды из колодца и водоемов, а также при неисправностях водопровода. Прямой контакт детей с животными или их испражнениями и передача от человека к человеку являются не менее важными механизмами передачи этой инфекции.
Классификация
Официальной классификации ГУС нет. По причинам возникновения выделяют инфекционные и неинфекционные формы (рис. 1). К инфекционным формам ГУС относят:
- ГУС, ассоциированный с шига-токсином (Sh. dysenteriae тип 1);
- ГУС, ассоциированный с микроорганизмами, секретирующими нейраминидазу (S. pneumoniae);
- ГУС, ассоциированный с ВИЧ-инфекцией, и др.
К неинфекционным формам относят идиопатический ГУС, наследственный ГУС (связанный с аномалиями ADAMTS-13), лекарственно-индуцированный ГУС (прием ингибиторов mTOR или ингибиторов VEGF) и другие формы.

Рисунок 1 | Классификация ГУС
Помимо вышеописанной классификации, ГУС можно отнести к первичным тромботическим микроангиопатиям (ТМА), этиология и патогенез которых установлены:
- ГУС, индуцированный инфекцией или ассоциированный с диареей (тГУС, ГУС-(D+));
- Атипичный ГУС, обусловленный генетическими нарушениями или изменениями иммунной системы, приводящими к патологии системы комплемента (аГУС, ГУС-(D–));
- Тромботическая тромбоцитопеническая пурпура (ТТП, болезнь Мошковица), связанная с аномалиями фермента ADAMTS-13 (врожденная или приобретенная).
Патогенез
Типичный ГУС
Основным фактором, инициирующим развитие тГУС, который обусловливает до 80 % от общего числа случаев заболевания, является энтерогеморрагическая кишечная палочка (E. coli, EHEC, серотип О157:Н7), синтезирующая шигаподобный токсин (веротоксин 1 и/или 2 типа). Данный штамм обладает высокой патогенностью для человека (для заражения достаточно 103 микроорганизмов), однако диарея развивается только в каждом 10-м случае (рис. 2).
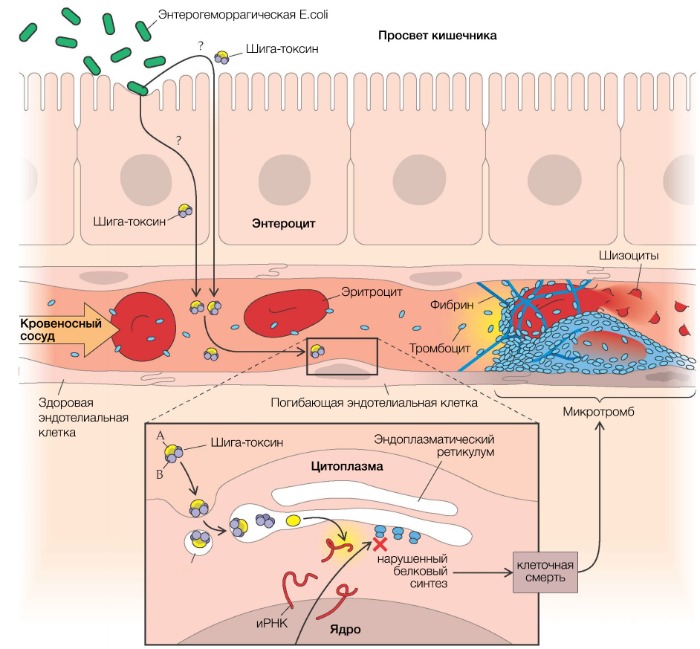
Рисунок 2 | Патогенез тГУС, ассоциированного с EHEC
После попадания E. coli в кишечник она связывается с ворсинками подвздошной кишки и эпителиальными клетками пейеровых бляшек при помощи специального белка, вызывая в конечном итоге гибель клеток с развитием диареи, переходящей в гемоколит (его возникновение связано с веротоксином, который способен повреждать сосуды слизистой оболочки кишечника). Шигаподобный токсин (SLT, Stx), высвобождающийся из кишечника, попадает в печень, где часть его метаболизируется, а другая часть попадает в системный кровоток, вызывая повреждение эндотелия органов-мишеней (легкие, почки, головной мозг).
SLT транспортируется в крови в основном нейтрофилами, но может перемещаться по системному кровотоку и при помощи моноцитов, тромбоцитов и/или их комплексов (липополисахарид кишечной палочки связывается с тромбоцитами, вызывая их активацию и агрегацию). За счет субъединицы В Stx имеет высокое сродство к мембраносвязанным гликосфинголипидам — Gb3/Gb4-рецепторам (в 100 раз выше, чем таковое с нейтрофилами).
В связи с этим повреждение эндотелия ярко выражено в мелких сосудах почек, но не в крупных сосудах других «возможных» органов-мишеней (экспрессия рецепторов Gb на мембранах эндотелиальных клеток почечных клубочков в 50 раз выше, чем в других тканях и органах). Помимо клеток эндотелия почечных клубочков, мезангиальных клеток, подоцитов, Gb-рецепторы в большом количестве синтезируются на мембранах нейронов и глиальных клеток. После проникновения Stx (за счет А субъединицы) внутрь клетки происходит блокирование синтеза белков путем инактивации рибосомальных субъединиц (60S) с последующим апоптозом этих клеток.
Помимо этого, ЛПС, концентрация которого в крови прямо коррелирует с таковой у шига-токсина, обусловливает повышенную продукцию провоспалительных цитокинов — интерлейкинов 1, 6, 8, а также фактора некроза опухоли-альфа (TNF-α). Те, в свою очередь, повышают экспрессию рецепторов на мембранах связывающих их моноцитов, приводя тем самым к более выраженному токсическому эффекту Stx.
Патогенез ГУС, вызванного Shigella dysenteriae 1 типа, схож с таковым у E. coli (рис. 3, 4, 5). Однако этот тип ГУС протекает тяжелее, чем ГУС, ассоциированный с шигаподобным токсином E. coli. Связано это, скорее всего, с липополисахаридным эндотоксином шигелл, который путем сложного взаимодействия с рецепторами TLR4 (на мембранах клеток) и NLR 1, 2 (Nod like receptors, расположены внутриклеточно) вызывает активацию сигнального пути NF-kB, что, в свою очередь, приводит к массивному выделению интерлейкина 8, являющегося мощным хемокином для нейтрофилов, макрофагов и лимфоцитов. Активированные нейтрофилы путем массивного выброса воспалительных цитокинов, помимо повышения секреции специфических рецепторов на мембранах эндотелиоцитов, вызывают активацию перекисного окисления липидов (ПОЛ), приводящего к повреждению не только эндотелия, но и эритроцитов, а также активацию лизосомальных ферментов, например, эластазы или α1-антитрипсина, которые также усугубляют эндотелиальное повреждение.
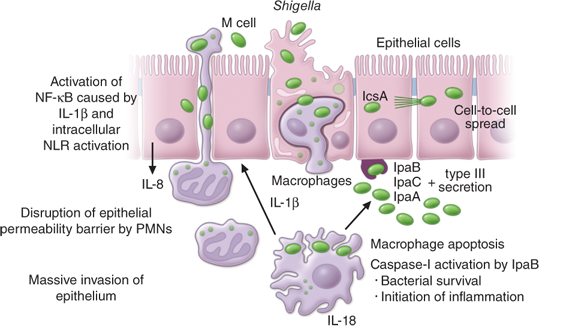
Рисунок 3 | Патогенез тГУС, ассоциированного с Shigella dysenteriae
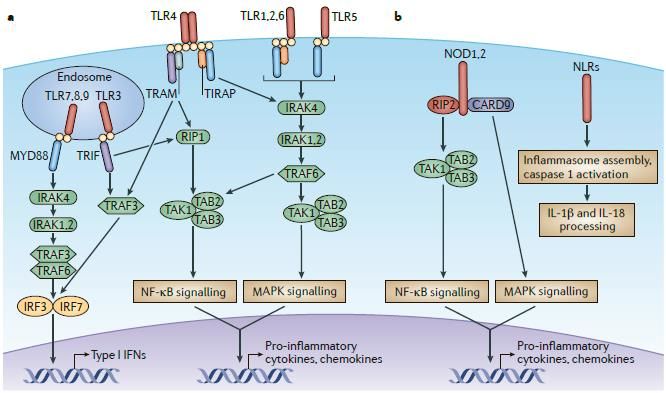
Рисунок 4 | Рецепторы TLR4 и NOD 1-2, через которые Shigella dysenteriae активирует сигнальный путь NF-kB
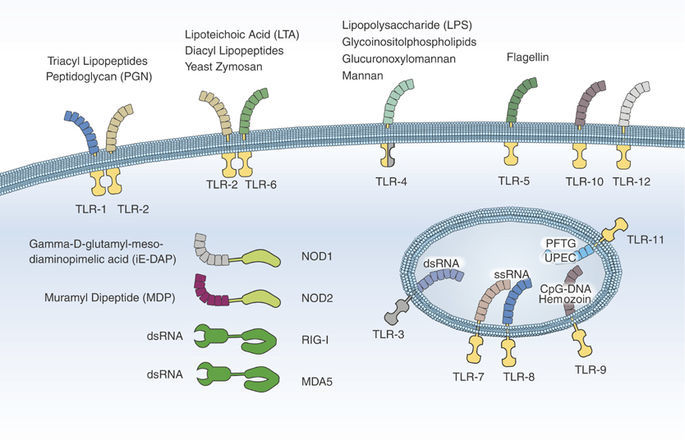
Рисунок 5 | Молекулярная основа патогенеза тГУС, ассоциированного с Shigella dysenteriae
ГУС, ассоциированный со Streptococcus pneumoniae, имеет несколько другой патогенез (рис. 6). Нейраминидаза S. pneumoniae атакует ацетилнейраминовую кислоту поверхности эндотелиальных клеток почечных клубочков, эритроцитов и тромбоцитов, обнажая при этом T-антиген (антиген Томсена-Фриденрайха). После «открытия» этих антигенов происходит усиление продукции Ig класса M, что, в свою очередь, приводит к агглютинации тромбоцитов и эритроцитов. По сути, эндотелий почечных клубочков повреждается как напрямую, так и иммуноопосредованно.
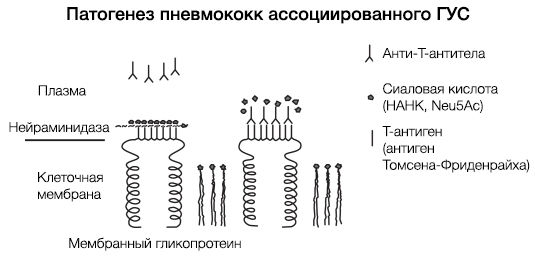
Рисунок 6 | Патогенез тГУС, ассоциированного со Streptococcus pneumoniae
В связи с этим происходит локальное изменение реологических свойств крови. Так называемый «shear stress» (связан с различной скоростью движения слоев крови), возрастающий при повреждении эндотелиальных клеток, помимо увеличения продукции NO (связано с раздражением механорецепторов), активирующего оксидативный стресс и инициирующего апоптоз эндотелиоцитов, также приводит и к агглютинации тромбоцитов. Основным звеном этой агглютинации является фактор Виллебранда (ФВ), который обычно накапливается в альфа-гранулах тромбоцитов и в тельцах Вайбеля-Паладе эндотелиальных клеток. Как говорилось выше, раздражение сосудистой стенки почечных клубочков путем воздействия на их стенку оксида азота, а также возрастающего касательного напряжения («shear stress») приводит к высвобождению ФВ, который способен поддерживать активацию и агрегацию тромбоцитов и тромбообразование в почечных клубочках.
Атипичный ГУС
Патогенез атипичного ГУС (аГУС) разительно отличается от тГУС. В его основе лежат мутации регуляторных белков системы комплемента (чаще всего аГУС ассоциирован с мутацией комплементарного фактора Н (CFH), на втором месте располагается мембранный кофакторный протеин (MCP), тройку замыкает комплементарный фактор I (CFI)).
При активации комплемента образуется C3-конвертаза, расщепляющая C3 на малый (С3а) и большой (C3b) фрагменты, который и опсонизируется на поверхности микробной клетки и формирует мембраноатакующий комплекс (МАК), состоящий из C5b, C6, C7, C8 и C9, что приводит к осмотическому лизису этой клетки. Для того, чтобы активированная система комплемента не уничтожила собственные клетки, на их поверхности расположены белки-регуляторы (DAF и CR1); помимо этого, часть таких белков синтезируется в печени и циркулирует в плазме крови в неактивном состоянии. К таким белкам относят комплементарный фактор H (CFH), фактор I (CFI) и мембранный кофакторный протеин, закрепленный на поверхности клеток (CD46). Фактор I, главный из вышеперечисленных факторов, расщепляет C3b и C4b. Фактор Н и CD46 являются кофакторами фактора комплемента I (рис. 7). Первый из них связывается с гликозаминогликанами собственных клеток организма, отсутствующими на мембранах бактериальных клеток, а также ингибирует активность C3-конвертазы. При мутации данных регуляторных белков происходит утрата защиты эндотелиальных клеток от повреждения конечными продуктами активации альтернативного пути комплемента (рис. 8).
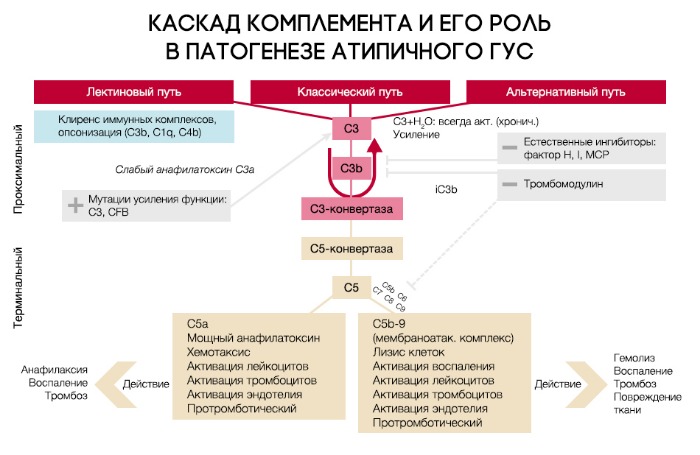 Рисунок 7 | Нормальная регуляция системы комплемента
Рисунок 7 | Нормальная регуляция системы комплемента
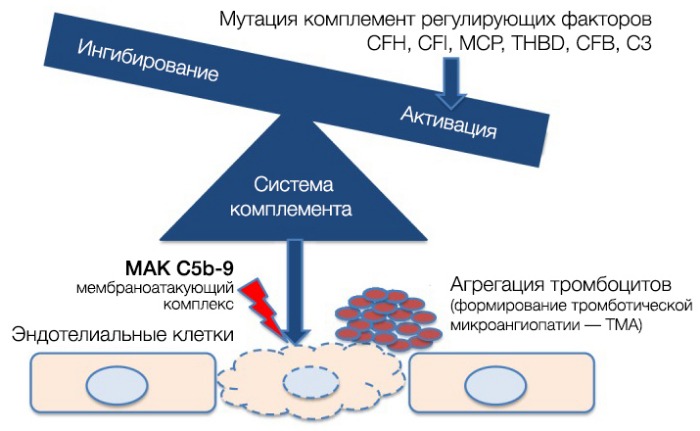
Рисунок 8 | Патогенез аГУС
Клиническая картина
В течении тГУС условно можно выделить 2 периода. Продромальный период характеризуется диареей, в ⅔ случаях диарею может сопровождать рвота. Гемоколит, характерный для тГУС, ассоциированного с шига-токсином, обычно развивается у каждого третьего больного через 2–3 дня после появления диареи.
Развернутая клиническая картина тГУС манифестирует в среднем на 5–6 день от начала диареи. Клиника крайне неспецифична. Бледность кожных покровов, общее недомогание, слабость, кожный геморрагический синдром в виде петехиальной сыпи или пурпуры, изменение цвета и уменьшение количества мочи после «кровавой» диареи должны насторожить врача в отношении тГУС (чаще всего диагностируется олигоанурическая стадия ОПН, требующая немедленного диализа).
Ввиду частой гиподиагностики адекватный контроль регидратации может отсутствовать, поэтому дополнительным признаком тГУС у детей можно считать артериальную гипертензию, которая отличается упорным течением и плохо поддается терапии. После восстановления нормального диуреза может отмечаться второй подъем АД, связанный с избыточной выработкой ренина.
Поражение ЦНС в виде генерализованных судорог, нарушения сознания вплоть до комы развивается в каждом четвертом случае и характеризует тяжесть течения тГУС.
Клиника аГУС имеет ряд особенностей!
Для аГУС характерно очень раннее начало (начиная с 2 месяцев при мутации CFI, c 6 месяцев при мутации CFH). Чаще всего аГУС манифестирует неспецифическими симптомами в виде общей слабости, недомогания без предшествующей этим симптомам диареи (при ее наличии затрудняется дифференциальный диагноз с тГУС). Возможно развитие гриппоподобного синдрома. У взрослых клиника может развиваться стерто, при этом классическая триада ГУС имеет слабую выраженность. Возможно развитие неполной триады без тромбоцитопении. Также для аГУС характерен семейный характер заболевания (в 25 % случаев имеется отягощенный наследственный анамнез). Большинство пациентов имеют выраженный отечный синдром вплоть до анасарки даже при отсутствии нефротического синдрома и ОПН. Также для больных с аГУС характерна АГ, связанная с перегрузкой объемом при манифестирующей ОПН или возникающая вследствие активации РААС, вызванной ишемией почечной ткани, обусловленной тромботической микроангиопатией. В отличие от тГУС, для атипичной формы характерно развитие кардиомиопатии с признаками острой СН. Описаны случаи развития острого панкреатита вплоть до панкреонекроза. В общем и целом клиника аГУС отличается яркой вариабельностью симптомов, что затрудняет ее своевременную диагностику (характерно рецидивирование).
Диагностика
Лабораторная диагностика как типичного, так и атипичного ГУС основана на выявлении признаков тромботической микроангиопатии:
- гемолитическая анемия — уровень гемоглобина ниже 90 г/л, выраженный ретикулоцитоз. Помимо этого, в крови могут появляться остатки эритроцитов — шизоциты (больше 1 %). Разрушение эритроцитов происходит из-за механического повреждения нитями фибрина при их прохождении через тромбированные сосуды почечных клубочков;
- тромбоцитопения ниже 150 тыс/мм3;
- выраженный лейкоцитоз выше 20 х 109/л; характеризует тяжесть ГУС.
Помимо этого, к неспецифическим признакам гемолиза эритроцитов можно отнести повышенный уровень ЛДГ, уменьшение уровня свободного гаптоглобина, гипербилирубинемию (за счет непрямой фракции). При проведении прямой пробы Кумбса результат будет отрицательным как у тГУС, так и аГУС.
Система диагностики ОПН, согласно критериям AKIN, основана на исследовании концентрации креатинина в сыворотке крови, СКФ, рассчитанной по формуле MDRD или CKD-EPI, а также на объеме мочи в течение суток. Для достоверной диагностики анурической стадии ОПН при подозрении на ГУС возможно выявление ранних признаков почечного повреждения (повышение уровня цистатина С, NGAL), а также повышения уровня калия крови выше 6 ммоль/л.
С целью подтверждения ГУС, связанного с шига-токсином, при наличии симптомов со стороны ЖКТ необходимо провести:
- посев кала на среду МакКонки для выявления серотипа E. coli O157:H7;
- определение шига-токсина в кале методом ПЦР или его выявление в сыворотке крови;
- возможно определение антител к липополисахариду эндемичного для данного региона серотипа E. coli.
Для исключения тромботической тромбоцитопенической пурпуры (ТТП) всем больным с характерной для ГУС клинической картиной необходимо определение активности ADAMTS-13 (менее 5 %). Для пациентов с аГУС типично снижение данного показателя, однако он в любом случае будет выше 10 % (в норме составляет 80–110 %).
Если при госпитализации больного в его анамнезе были выявлены предшествующие тромботические микроангиопатии, необходимо исследовать кровь на содержание С3 и С4 компонентов комплемента, а также на аутоантитела к фактору Н (анти-FH-антитела). Помимо этого, необходимо проведение дифференциальной диагностики со системными заболеваниями соединительной ткани (рис. 9). Развитие характерного симптомокомплекса во время беременности требует исключения специфической акушерской патологии.

Рисунок 9 | Дифференциальная диагностика ГУС
Лечение
Относительно специфическое лечение разработано только для атипичной формы ГУС. В настоящее время единственным допущенным до применения ингибитором системы комплемента является экулизумаб (рекомбинантное моноклональное антитело против компонента комплемента С5). Данный препарат блокирует расщепление С5 компонента комплемента (С5а — провоспалительный, C5b — протромботический компонент) и формирование на мембране собственных клеток МАК C5b-9.
Клинические испытания показали, что в профилактике и лечении аГУС экулизумаб оказался более эффективен, чем плазмаферез. Однако наилучшей схемой лечения считается введение препарата на фоне проведения плазмафереза, так как последний удаляет часть препарата из циркуляции, тем самым предотвращая развитие побочных реакций.
Введение свежезамороженной плазмы (СЗП) не предотвращает развитие терминальных стадий ХБП у больных с аГУС. Ее использование оправдано ввиду того, что СЗП является источником нормальных комплементарных факторов CFH и CFI. Как и в ситуации с экулизумабом, введение СЗП лучше сочетать с плазмаферезом (удаляются мутантные комплементарные факторы и анти-CFH антитела; удаление части плазмы предотвращает гиперволемию и следующую за этим острую сердечную недостаточность). Ввиду генетической детерминированности аГУС высок риск развития рецидива. Это, в свою очередь, требует проведение адекватной профилактической терапии, включающей в себя санацию очагов хронической инфекции, а также проведение своевременной вакцинопрофилактики. Трансплантация почки как метод лечения аГУС не имеет на данный момент широкой доказательной базы (описано всего 3 случая пересадки). Риск возврата аГУС сразу после трансплантации чрезвычайно высок, особенно у пациентов с мутацией комплементарного фактора Н.
Консервативное лечение тГУС заключается в проведении корректной регидратационной терапии. Как говорилось выше, неспецифичность клинической картины обусловливает высокий процент гиподиагностики, в связи с чем следующая за этим попытка коррекции водно-электролитного баланса оказывает отрицательный эффект (например, перегрузка объемом приводит в итоге к развитию острой сердечной недостаточности и активации РААС (гиперренинемии) — формируется резистентная к проводимой терапии артериальная гипертензия. Использование петлевых диуретиков, например, фуросемида, не оправдано; предпочтение стоит отдавать гемодиализу (на фоне гиперкалиемии или метаболического ацидоза) ввиду того, что диализ чаще всего начинается в олигоанурической стадии ОПН. При развитии анемии тяжелой степени (Hb ниже 70 г/л) показано переливание эритроцитарной массы. Антибиотики не являются основным компонентом терапии тГУС. Однако раннее назначение цефалоспоринов III поколения или фторхинолонов снижает риск развития тГУС, ассоциированного с S. dysenteriae типа 1.
Источники:
- Gasser C. et al. Hemolytic-uremic syndrome: bilateral necrosis of the renal cortex in acute acquired hemolytic anemia //Schweizerische medizinische Wochenschrift. – 1955. – Т. 85. – №. 38-39. – С. 905.
- Цыгин А. Н. и др. Федеральные клинические рекомендации по оказанию помощи детям с гемолитико-уремическим синдромом //Педиатрическая фармакология. – 2015. – Т. 12. – №. 4.
- Mayer C. L. et al. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals //Toxins. – 2012. – Т. 4. – №. 11. – С. 1261-1287.
- Boyer O., Niaudet P. Hemolytic uremic syndrome: new developments in pathogenesis and treatment //International journal of nephrology. – 2011. – Т. 2011.
- Taylor C. M. Enterohaemorrhagic Escherichia coli and Shigella dysenteriae type 1-induced haemolytic uraemic syndrome //Pediatric Nephrology. – 2008. – Т. 23. – №. 9. – С. 1425.
- Oliver J. W. et al. Pneumococcal induced T-activation with resultant thrombotic microangiopathy //Clinical Medicine Insights: Pathology. – 2010. – Т. 3. – С. CPath. S670.
- Чубуков Ж. А. Фактор Виллебранда и дисфункция эндотелия при стрессе //Проблемы здоровья и экологии. – 2012. – №. 2 (32).
- Fremeaux-Bacchi V. Pathophysiology of atypical hemolytic uremic syndrome. Ten years of progress, from laboratory to patient //Biologie aujourd'hui. – 2013. – Т. 207. – №. 4. – С. 231-240.
- Zhang K. et al. Atypical hemolytic uremic syndrome: a brief review //Hematology reports. – 2017. – Т. 9. – №. 2.
- Salvadori M., Bertoni E. Update on hemolytic uremic syndrome: diagnostic and therapeutic recommendations //World journal of nephrology. – 2013. – Т. 2. – №. 3. – С. 56.
- Козловская Н. Л. и др. Клинические рекомендации по диагностике и лечению атипичного гемолитико-уремического синдрома //Нефрология и диализ. – 2015. – Т. 17. – №. 3. – С. 242-264.
- Cheong H. I. et al. Clinical practice guidelines for the management of atypical hemolytic uremic syndrome in Korea //Journal of Korean medical science. – 2016. – Т. 31. – №. 10. – С. 1516-1528.
- Kaplan B. S. et al. Current treatment of atypical hemolytic uremic syndrome //Intractable & rare diseases research. – 2014. – Т. 3. – №. 2. – С. 34-45.
- ВКонтакте
- РћРТвЂВВВВВВВВнокласснРСвЂВВВВВВВВРєРСвЂВВВВВВВВ
- Telegram
