
Нейродегенерация с отложением железа в головном мозге

В 1922 году два ученых из Германии, Юлиус Галлеворден и Гуго Шпатц, занялись изучением следующего клинического случая: в семье из 12 человек у пяти сестер наблюдались прогрессирующая деменция и дизартрия. Данные аутопсии показали, что в многочисленных областях головного мозга ткани были окрашены в коричневый цвет. В частности, интерес представляла окраска в области чёрной субстанции и базальных ядер. Описанное заболевание получило название синдрома Галлевордена — Шпатца и было под ним известно вплоть до недавнего времени, пока не была установлена роль исследователей в проведении экспериментов над заключенными в нацистской Германии, после чего эпоним сменили на нейродегенерацию с отложением железа в головном мозге (Neurodegeneration with brain iron accumulation, NBIA).
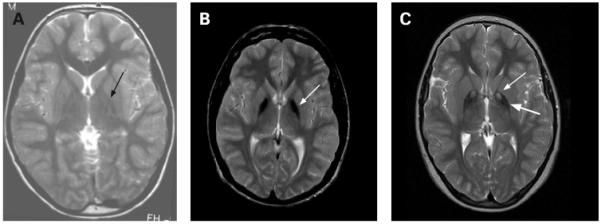
Рисунок 1. Т2-взвешенные снимки. А - здоровый пацент. В - идиопатическая NBIA, стрелкой обозначена зона гипоинтенсивности в медиальном бледном шаре. С - PKAN, регион гиперинтенсивности (тонкая стрелка) окружён гипоинтенсивной зоной (толстая стрелка), так же в медиальном бледном шаре, т.н "глаз тигра" - типичное для PKAN распределение железа.
До 2001 года синдромом Галлевордена — Шпатца называли все формы NBIA, однако после открытия первого NBIA-ассоциированного гена от этого пришлось отказаться. Изученный ген исследователи связали с пантотенат-киназно связанной нейродегенерацией (Pantothenate kinase-associated neurodegeneration, PKAN) — самой распространенной формой нейродегенерации с отложением железа. В течение последующих лет были открыты и другие генные мутации и фенотипы нейродегенерации: в 2006 году была обнаружена мутация гена PLA2G6, кодирующего фосфолипазу А2, приводящая к возникновению PLA2G6-ассоциированной нейродегенерации (PLAN), в 2011 — ген C19orf12 был идентифицирован как ответственный за нейродегенерацию, ассоциированную с белками митохондриальной мембраны (MPAN). Еще более недавно в спектр NBIA включили еще одно заболевание: бета-пропеллерный белок-ассоциированную нейродегенерацию (BPAN). Вместе эти четыре подтипа нейродегенерации с отложением железа являются наиболее часто встречающимися и диагностируемыми (более чем 62 % от всех случаев).
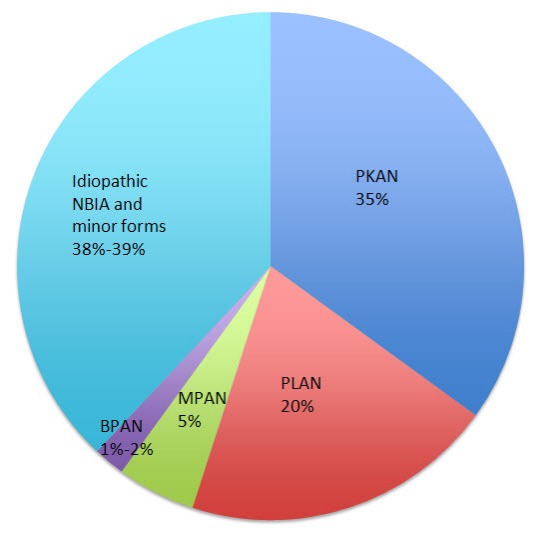
Рисунок 2. Частота встречаемости подтипов NBIA.
В спектр также были включены еще 6 более редких подтипов: нейроферритинопатия, ацерулоплазминемия, болезнь Куфора — Ракеба, нейродегенерация, связанная с гидроксилазой жирных кислот (FAHN), синдром Вудхауза — Сакати и COASY-протеин-ассоциированная нейродегенерация (CoPAN). Большая часть заболеваний, объединенных термином NBIA наследуется аутосомно-рецессивно, за исключением BPAN и нейроферритинопатии, которые наследуются Х-сцепленно доминантно и аутосомно-доминантно соответственно.
В случаях, когда этиология заболевания не может быть установлена, пациенту диагностируют идиопатическую нейродегенерацию с накоплением железа. Особенно усложняется постановка диагноза в том случае, если пациент — единственный в семье человек с заболеванием. Даже если есть вероятность наследования болезни по аутосомно-рецессивному типу, это следует подтвердить при помощи проведения молекулярно-генетического исследования.
Течение заболевания очень сильно варьирует: манифест может случиться в любом возрасте, начиная от младенчества. Прогрессируют симптомы в ряде случаев стремительно, а в ряде — медленно, с длительными периодами стабильности. Конкретно симптоматика определяется генетическими механизмами, вызвавшими нейродегенерацию, но факторы, влияющие на тяжесть и скорость прогрессирования все еще остаются неизвестны.
Характерными признаками NBIA являются прогрессирующая дистония, дизартрия, спастичность, паркинсонизм и отставание в интеллектуальном развитии. Часто симптомы сопровождаются дегенерацией сетчатки и атрофией зрительного нерва. Среди нейропатологических признаков отмечают не только накопление железа, но еще и образование аксональных сфероидов в ЦНС и иногда в периферических нервах.
Самым распространенным типом NBIA является PKAN (35–50 % случаев). Фенотипически выделяют классическую PKAN, для которой характерен ранний манифест и быстрое развитие, и атипичную, с более поздним началом, примерно в 13–14 лет и не таким стремительным ухудшением. Для детей с пантотенат-киназно связанной нейродегенерацией характерно появление проблем с походкой в возрасте около трех лет и затем прогрессирование дистонии, ригидности, дизартрии, гиперрефлексии, спастичности и рефлекса Бабинского. Последние три симптома «обязаны» своим наличием поражению кортикоспинального тракта. У лиц с атипичной формой этого подтипа нейродегенерации чаще заболевание начинает проявляться с проблем с речью; для более поздно манифестирующей формы характерны психиатрические симптомы (депрессия, эмоциональная лабильность, импульсивность, ОКР).
Существует корреляция между скоростью прогрессирования болезни и возрастом появления первых симпотомов. Несмотря на это, были зарегистрированы случаи очень быстрого ухудшения состояния у лиц с манифестом в раннем подростковом возрасте вплоть до смерти до достижения 20 лет. Напротив, есть и пациенты, дожившие до 50 лет, несмотря на проявление первых симптомов в возрасте до 10.
По мере ухудшения состояния пациентам часто требуется кормление через гастральный зонд из-за развития дисфагии. Гастроэзофагеальный рефлюкс и запоры становятся хронической проблемой для больных на поздних стадиях заболевания. Обычно причиной смерти становятся вторичные осложнения, например, аспирационная пневмония и истощение.
Исторически, NBIA ассоциировалась исследователями с нарушениями интеллекта. В исследовании с 16 пациентами с PKAN оба из проведенных тестирований выявили отрицательную корреляцию уровня IQ с тяжестью заболевания. То есть испытуемые с самыми тяжелыми проявлениями хуже всего справились с тестами.
В случае если речь идет о других формах заболевания из спектра, постановка диагноза может оказаться гораздо более затруднительной, чем при PKAN. В этом случае на помощь приходит МРТ, подходящая для первичного определения формы нейродегенерации, тем самым наталкивая врача на выбор правильного молекулярно-генетического исследования. МРТ головного мозга является стандартным методом исследования для описываемой группы заболеваний. Более новые нейровизуализационные технологии, например, магнитно-резонансная спектроскопия (МРС), предположительно, тоже могут оказаться полезными для постановки диагноза, однако на данный момент это достоверно неизвестно.
По определению в базальных ганглиях людей с NBIA определяется ненормально высокое количество железа. Обычно эти зоны определяются как гипоинтенсивные участки бледного шара и ретикулярной части черной субстанции на Т2-взвешенных снимках. На Т1-взвешенных снимках эти области изоинтенсивны, что помогает отличить их от отложений кальция и прочих изменений. При идиопатической нейродегенерации с накоплением железа участки его аккумуляции могут встречаться в красном ядре, зубчатом ядре, скорлупе и хвостатом ядре. После постановки диагноза проводить повторные исследования при помощи МРТ не имеет смысла. Следует помнить, что порой диагностика с целью выявления конкретной формы NBIA может занять несколько лет. Например, в случае болезни Куфора — Ракеба или PLAN накопление железа может произойти только в ряде случаев.
Как фармакологические, так и хирургические вмешательства могут быть направлены только на паллиативную терапию в связи с этиологией нейродегенераций. Многие из этих вмешательств предоставляют лишь временное улучшение, поэтому для поддержания максимального высокого качества жизни пациента необходимы периодические корректировки хода лечения. Наиболее эффективными препаратами для снятия дистонии и спастичности являются баклофен и тригексифенидил. Как правило, пациенты с PKAN не реагируют на L-дофа, в то время, как пациенты с другими формами NBIA и паркинсонизмом в некоторых случаях реагируют на L-дофа, особенно в редких случаях, характеризующихся ранними задержками развития и проявлением дистонии уже во взрослом возрасте. Ботулотоксин может быть полезен для пациентов, чье качество жизни может быть улучшено терапией конкретной части тела. Например, инъекции в мышцы лица способствуют улучшению речи и способности к самостоятельному питанию.
Источники:
1. https://goo.gl/9W749H
2. Limongi J. C. P, Neurodegeneration with brain iron accumulation. Arq Neuropsiquiatr 2016;74(7):517-518
3. https://goo.gl/Q2xD1E
4. Gregory, A.,Polster, B. J.,Hayflick, S. J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation / A. Gregory, B. J. Polster, S. J. Hayflick // J Med Genet. - 2009. - Т. 46. - №2. - С. 73-80
