
Инфламмасома — белковый комплекс, зачинщик воспаления
Воспаление — это защитный иммунный ответ, эволюционно установленный врожденной иммунной системой в ответ на вредоносные стимулы, такие как: патогены, мертвые клетки или раздражители, и оно жестко регулируется хозяином. Недостаточное воспаление может привести к хроническому течению инфекции, в то время как чрезмерное воспаление может привести к системным воспалительным заболеваниям.
Функция врожденного иммунитета зависит от распознавания ассоциированных с патогенами молекулярных структур (PAMP-pathogen-associated molecular patterns), полученных из патогенных микроорганизмов, и от связанных c дистресс-ассоциированными молекулярными структурами (DAMP), которые индуцируются в результате эндогенного стресса путем распознавания образов посредством зашифрованных зародышевых линий рецепторов (PRR-pattern-recognition receptors). Активация PRR с помощью PAMP или DAMPs триггеров посылает нисходящие сигнальные каскады и приводит к продукции интерферонов типа I (интерферон-α и интерферон-β) и провоспалительных цитокинов. Следует отметить, что воспаление, вызванное DAMP, которые особенно важны в воспалительных заболеваниях, называется стерильным, если это происходит в отсутствие каких-либо чужеродных патогенов.
Активация инфламмасом является ключевой функцией, опосредованной врожденной иммунной системой, а недавние успехи значительно увеличили понимание макромолекулярной активации воспалительных заболеваний. Несколько семейств PRR являются важными компонентами комплекса инфламмасом, включая нуклеотид-связывающий домен, лейцин-насыщенные повторы. При обнаружении определенных раздражителей соответствующий NLR или AIM2 может олигомеризоваться в каспаза-1-активирующий каркас. Активная каспаза-1 впоследствии участвует в расщеплении провоспалительного семейства цитокинов IL-1 в их биологически активные формы IL-1β и IL-18 и вызывает пироптоз — тип воспалительной гибели клеток.
Механизмы активации инфламмасом
NLRP3 inflammasome
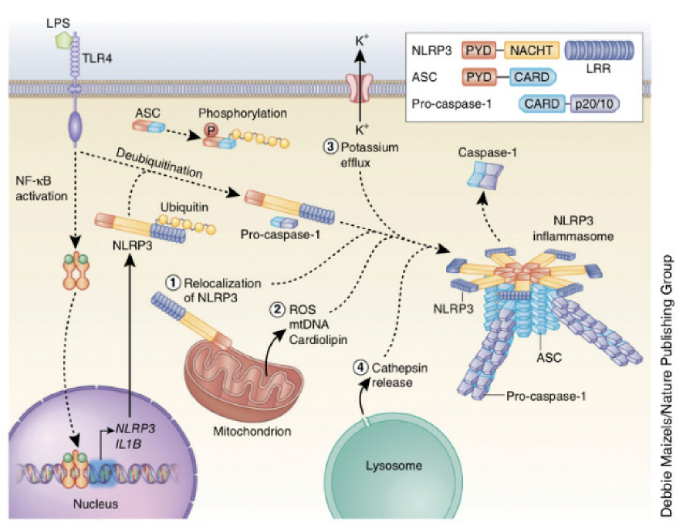
NLRP3 инфламмасома активируется в ответ на широкий набор стимулов, и этот факт приводит к теории, которая предполагает, что агонисты разной природы вызывают подобные процессы, ощущаемые NLRP3. Механизмы активации NLRP3, поддерживаемые большинством исследований, включают в себя: отток калия из клетки, образование митохондриальных активных видов кислорода (ROS — reactive oxygen species), транслокация NLRP3 в митохондрии, выделение митохондриальной ДНК, или кардиолипина, и выделение катепсинов в цитозоль после лизосомальной дестабилизации. Однако не все эти события индуцируются агонистами NLRP3, поэтому точный механизм активации NLRP3 все еще стоит под вопросом.
В дополнение к этому увеличение концентрации внутриклеточного кальция может активировать NLRP3 инфламмасому, но это не является обязательным требованием для всех агонистов NLRP3. Хотя многие опубликованные исследования поддерживают участие лизосомальных катепсинов, протеаз, которые расщепляют внутренние белки, при активации NLRP3 инфламмасом, важно отметить, что не обошлось без разногласий. В большинстве типов клеток NLRP3 должен быть инициирован, а примером такого инициирующего события является связывание липополисахарида (LPS) с TLR4. Известно, что инициация увеличивает клеточную экспрессию NLRP3 через cигнальный каскад NF-κB. Однажды возбудившись, NLRP3 может воздействовать на раздражители и собирать NLRP3 инфламмасому. Кроме того, ASC (apoptosis-associated speck-like protein containing a CARD) должен быть линейно убиквитирован (прим. —убиквитинирование — это посттрансляционное присоединение ферментами убиквитинлигазами одного или нескольких мономеров убиквитина с помощью ковалентной связи к боковым аминогруппам белка-мишени) для сборки NLRP3 инфламмасомы.
Существующие стимулы распознаются как агонисты NLRP3, которые индуцируют образование NLRP3 инфламмасомы, включающие в себя: АТФ, порообразующие токсины, кристаллические вещества, нуклеиновые кислоты, гиалуроновые и грибковые, бактериальные или вирусные патогены. Эти стимулы могут обнаружиться во время инфекции, или продуцироваться патогенами, или же отделиться от поврежденной клетки-хозяина. Кроме того, патологические состояния в организме могут способствовать формированию этих раздражителей в отсутствие инфекции; примером является образование воспалительных форм кристаллов холестерола. Недавние исследования показали, что NLRP3 NBD олигомеризует NLRP3 PYD, который служит в качестве каркаса для синтеза белков ASC через PYD-PYD взаимодействия.
Это приводит к преобразованию ASC в прион-подобную форму и преобразует короткие цепи в активные длинные нити ASC, которые имеют решающее значение в активации инфламмасомы. Pro-caspase-1 затем взаимодействует с ASC как CARD-CARD и формирует свои собственные прион-подобные нити, которые отделяются от нитей ASC. Близость белков прокаспазы-1 затем индуцирует аутопротеолитическое созревание прокаспазы-1 в активную каспазу-1. Кроме того, увеличение числа доказательств определило решающую роль для каспазы-8 при активации инфламмасомы и процессинга (созревания) про-IL-1β. Каспаза-8 представляет собой проапоптотическую протеазу, которая инициирует внешний апоптозный путь в ответ на внешние раздражители, такие как FasL и TNF (фактор некроза опухоли), и защищает от воспалительной формы клеточной смерти — некроптоза.
В настоящее время также признано, что каспаза-8 требуется для транскрипционной инициации и активации типичных и нетипичных NLRP3 инфламмасом в ответ на патогенные стимулы и лиганды, стимулирующие различные TLRs (Toll-like receptors). Таким образом, воспалительные заболевания, в которых образуются TLR-лиганды, могут привести к каспазой-8-опосредованному NLRP3 инициации или активации инфламмасомы. Важно отметить: каспаза-8 также имеет известную роль в активации NLRC4 и AIM2 инфламмасом, и даже было показано, что непосредственно влияет на процессинг про-IL-1β в комплексе неканонической каспаза-8-инфламмасома, вызванным связыванием некоторых внеклеточных патогенов в dectin-1.
AIM2 inflammasome
Не-NLR AIM2 может также образовывать каспаза-1-содержащую инфламмасому, но, в отличие от NLR, домен HIN-200 AIM2 может напрямую связывать его стимул, цитозольную dsDNA(двуцепочеченую ДНК), которые могут встречаться в цитозоле во время патогенной инфекции. Автоингибирующая конформация AIM2 создается взаимодействием из двух его доменов и освобождена от сахарофосфатного остова dsDNA. AIM2 не может взаимодействовать с ASC, если автоингибирование не прекращено, и, таким образом, AIM2 поддерживает себя в неактивном состоянии, пока его лиганд не свяжется. Интересно, что AIM2, похоже, не распознает определенную последовательность или структуры dsDNA, но вместо этого требуется цепочка dsDNA длиною не менее 80 пар оснований для оптимальной активации инфламмасомы. Аналогично NLRP3, олигомеризованные AIM2 собирают вокруг ядра ASC через PYD-PYD взаимодействия и преобразуют ASC в свою прионную форму, что приводит к образованию длинных PYD-PYD ASC нитей. Недавно была показана нетипичная инфламмасома AIM2 для защиты от Francisella novicida. F. novicida обнаруживается посредством cGAS и STING, индуцируя экспрессию транскрипционного фактора IRF1. IRF1 увеличивает экспрессию гуанилатсвязывающих белков, которые увеличивают внутриклеточное уничтожение бактерии. Этот процесс вызывает высвобождение dsDNA в цитозоль и индуцирует активацию AIM2 инфламмасомы.
Роль инфламмасомы в различных заболеваниях (например, болезнь Альцгеймера AD)
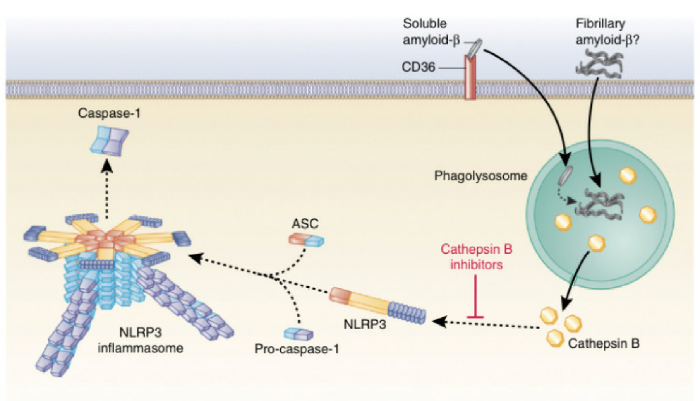
Накопление амилоидного бета белка в головном мозге является главной характеристикой AD. Амилоид-β-пептид регулярно формируется в мозговой ткани путем расщепления белка амилоидного предшественника, но он может образовывать прион-подобные неправильно упакованные олигомеры. Бета-амилоид был первой молекулой, ассоциированной с нейродегенеративными заболеваниями, которые были обнаружены при активации NLRP3 инфламмасомы мыши, в результате чего продуцировался IL-1β. Фибриллярный амилоид-β индуцирует NLRP3 инфламмасому ,зависимую от активации каспазы-1 через механизм, зависящий от эндосомального разрыва и высвобождения катепсина В в LPS-инициированные макрофаги мыши. Интересно, что введение ингибиторов катепсина В значительно улучшило память и уменьшило накопление амилоидного белка, предлагая потенциальный терапевтический подход к лечению AD, в котором мишенью является инфламмасома.
Источники
- Guo H., Callaway J. B., Ting J. P. Y. Inflammasomes: mechanism of action, role in disease, and therapeutics //Nature medicine. – 2015. – Т. 21. – №. 7. – С. 677.
- Kim J. K. et al. Negative regulators and their mechanisms in NLRP3 inflammasome activation and signaling //Immunology and cell biology. – 2017. – Т. 95. – №. 7. – С. 584-592.
- Кувачева Н. В. и др. Формирование инфламмасом: новые механизмы регуляции межклеточных взаимодействий и секреторной активности клеток //Сибирское медицинское обозрение. – 2013. – №. 5.
