
Описано взаимодействие белков, важное для гибели клеток
Австралийские исследователи показали, что белок VDAC2 активирует белок BAX, участвующий в апоптозе. Взаимодействие белков также важно для гибели раковых клеток под действием химиотерапии.
Апоптоз — распространенный способ клеточного самоубийства, который зачастую ингибируется в опухолевых клетках. Для разработки противоопухолевых препаратов важно понимание молекулярных механизмов его реализации. Так в работе, опубликованной в Nature Communications, объясняется необходимость взаимодействия белков VDAC2 и BAX для осуществления апоптоза.
В процессе запуска клеточной гибели, цитоплазматические белки BAK и BAX связываются с наружной мембраной митохондрий, формируют пору, через которую выходит цитохром C, запускающий дальнейшие проапоптотические реакции. Генетический скриниг с использованием технологии CRISPR/Cas9 показал, что VDAC2 (Voltage-dependent anion-selective channel protein 2) — один из ионных каналов внешней мембраны митохондрий — необходим для функционирования белка BAX, но не BAK. Исследователи определили, что формирование комплекса BAX с VDAC2 нужно для посадки BAX на мембрану митохондрий, в то время как BAK может садится на митохондрии самостоятельно.
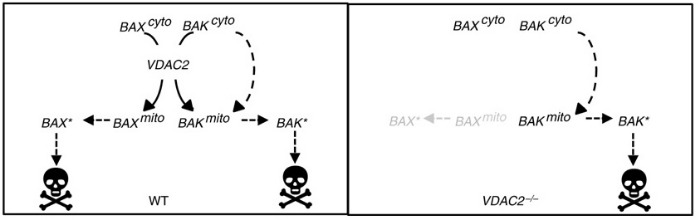
Некоторые противоопухолевые препараты запускают апоптоз в раковых клетках. В работе рассмотрели воздействие таких препаратов на клеточные линии раковых клеток и на опухоли in vivo. Эксперименты подтвердили, что для гибели таких клеток посредством BAX белок VDAC2 также необходим.
В случае предотвращения развития опухоли посредством апоптоза «ошибочных» клеток, VDAC2 является ключевым медиатором запуска BAX-зависимого апоптоза. Это было показано с помощью инъекции мышам протоонкогенного белка c-MYC для развития у них острого миелоидного лейкоза и выключения работы белков BAX, BAK и VDAC2.
Таким образом, VDAC2 способствует реализации BAX-зависимого апоптоза. Это взаимодействие можно использовать для разработки терапии в тех случаях, когда BAX чаще инициирует апоптоз, чем BAK. Например, в дифференцированных нейронах мало BAK, поэтому терапевтическое нарушение взаимодействия VDAC2 и BAX предотвратит патологический апоптоз этих клеток при травматических или ишемических повреждениях мозга. Исследование также актуально в случае лечения хронического лимфолейкоза препаратом Венетоклакс (Roberts et al., 2016), ингибирующего работу антиапоптотического белка BCL-2 и запускающего апоптоз раковых клеток. Иногда развивается резистентность к препарату, что может быть обусловлено мутациями в гене белка VDAC2 и последующим нарушением апоптоза.
Источники
Исследование: https://www.nature.com/articles/s41467-018-07309-4#ref-CR1
Паблик релиз: https://www.sciencedaily.com/releases/2018/11/181126123315.htm
