
Оптикомиелит Девика
Оптикомиелит (болезнь Девика) долгое время вызывал споры. Стоит ли считать это заболевание подтипом рассеянного склероза (РС) или самостоятельной нозологической формой? Открытие специфических антител к каналам аквапорина-4 (AQP4), которые широко представлены в отростках астроцитов, позволило окончательно разделить эти заболевания. В 2006 году произошел пересмотр критериев оптикомиелита и спектра оптикомиелит-ассоциированных расстройств (neuromyelitis optica spectrum disorders или NMOSD), по результатам которого для постановки диагноза требуется обнаружение антител к AQP4. Эти критерии и критерии Макдональда, используемые для диагностики РС, делают акцент на данные нейровизуализации. Однако часто бывает, что результаты МРТ не позволяют различить эти состояния, по этой причине NMOSD часто принимаются за РС, что может привести к ятрогении, т. к. NMOSD требует длительной иммуносупрессивной терапии для предотвращения рецидивов, а терапия РС среди прочего включает в себя натализумаб и интерферон-β, которые могут привести к обострению NMOSD. [2]
Оптикомиелит манифестирует, как правило, невритом зрительного нерва и продольно распространенным поперечным миелитом (longitudinal extensive transverse myelitis — LETM), хотя последние исследования говорят о вовлеченности циркумвентрикулярных органов и скелетных мышц. Оптикомиелит протекает с рецидивами, часто ассоциирован с другими аутоиммунными заболеваниями и иногда является составляющей паранеопластического синдрома. Специфичность исследования антител к AQP4 приближается к 100 %. Клинические, иммуногистопатологические и физиологические данные in vitro подтверждают, что эти аутоантитела являются ключевыми в патогенезе оптикомиелита. Последние исследования на животных дают ограниченные гистопатологические характеристики при отсутствии клинического дефицита. Недавно описанные антитела к миелин-олигодендроглиоцитарному гликопротеину у небольшой группы пациентов с фенотипом NMOSD и отсутствием AQP4-IgG дают основания для дальнейшего выделения новых нозологических форм.
Оптикомиелит Девика — это воспалительное аутоиммунное заболевание нервной системы, которое проявляется продольным распространенным поперечным миелитом (LETM) и невритом зрительного нерва. Открытие сывороточных специфических иммуноглобулинов G к каналам аквапорина-4 позволило унифицировать сам оптикомиелит и спектр расстройств, связанных с ним (NMOSD), а также достоверно отличить его от РС. Однако остается неясной взаимосвязь идиопатического оптиконеврита и поперечного миелита в рамках AQP4-IgG-серонегативного оптикомиелита с наличием альтернативных аутоантител, для которых поражение ЦНС еще не определено иммуногистопатологическими методами.
NMOSD не оценивался по таким признакам, как расовая или этническая принадлежность в различных популяциях, однако в западных странах выявляется непропорциональное распределение среди небелых пациентов. В Азии NMOSD диагностируется несколько чаще, чем РС. Женщины болеют чаще, по разным оценкам, от 3,6:1 до 10,4:1,9. Заболевание манифестирует в возрасте 35–45 лет, однако 18 % случаев приходится на детский или пожилой возраст. Хотя большая часть случаев спорадические, редкие случаи семейного серопозитивного AQP4-IgG-оптикомиелита с классическим фенотипом указывают на генетическую предрасположенность.
Клиника
В таблице 1 суммированы критерии оптикомиелита 2006 года. Девик в 1894 году впервые описал женщину с задержкой мочи, параплегией, двусторонней слепотой и отеком соска зрительного нерва. Аутопсия выявила острый миелит и билатеральный неврит зрительных нервов. Одновременный двусторонний оптиконеврит и поперечный миелит — редкая современная клиническая картина оптикомиелита. Такие случаи обычно протекают монофазно, одинаково часто встречаются у мужчин и женщин и являются AQP4-IgG-серонегативными. Таким образом, они не могут быть тождественны аутоиммунной каналопатии AQP4. Более длительный интервал между начальными клиническими симптомами и поздними проявлениями, манифестация в старшем возрасте и женский пол предсказывает рецидивирующее течение оптикомиелита. Хотя, в отличие от РС, вторично прогрессирующее течение не характерно.
Одно- или двусторонний неврит зрительных нервов отличается более тяжелым течением при NMOSD, чем при РС, с полным восстановлением только в 32 % случаев; поперечный миелит (обычно продольно распространенный) с полным клиническим восстановлением — только в 17 % случаев (рис. 1). Небольшие очаги в спинном мозге выявляются на МРТ в 14 % случаев при манифестации, а в случае рецидива 92 % очагов уже являются продольно распространенными. Поражение мозгового конуса и пояснично-крестцовых отделов не характерно.
 Таблица 1 ❘ Критерии оптикомиелита Девика, 2006
Таблица 1 ❘ Критерии оптикомиелита Девика, 2006
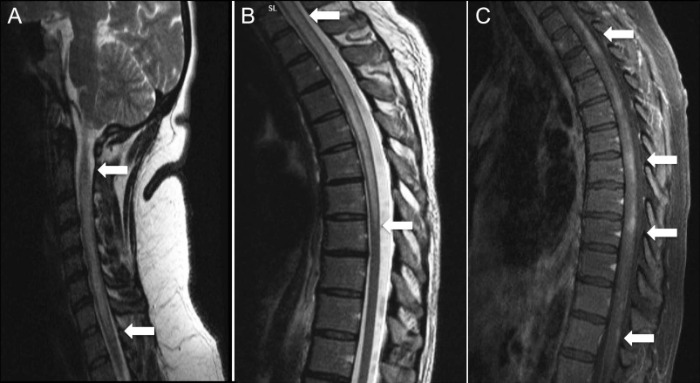 Рисунок 1 ❘ Продольно распространенный поперечный миелит (LETM) шейного (изобр. А, Т2-ВИ) и шейно-грудного (изобр. В, Т2-ВИ) отделов позвоночника с негомогенным контрастным усилением (изобр. С, Т1-ВИ после контрастирования) у AQP4-IgG-позитивных пациентов. [1]
Рисунок 1 ❘ Продольно распространенный поперечный миелит (LETM) шейного (изобр. А, Т2-ВИ) и шейно-грудного (изобр. В, Т2-ВИ) отделов позвоночника с негомогенным контрастным усилением (изобр. С, Т1-ВИ после контрастирования) у AQP4-IgG-позитивных пациентов. [1]
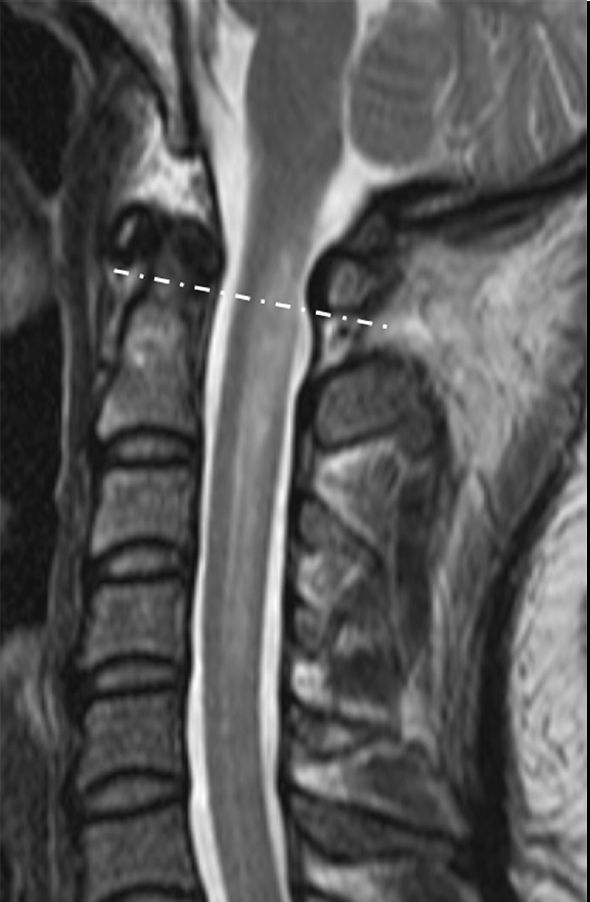 Рисунок 2 ❘ МРТ Т2-ВИ женщины 36 лет с LETM. Очаг поражения распространяется за пределы спинного мозга (пунктирная линия). [3]
Рисунок 2 ❘ МРТ Т2-ВИ женщины 36 лет с LETM. Очаг поражения распространяется за пределы спинного мозга (пунктирная линия). [3]
Спектр оптикомиелит-ассоциированных расстройств (NMOSD)
Наличие AQP4-IgG объединяет пациентов с малыми формами оптикомиелита, которые не удовлетворяют в полной мере критериям диагноза. Клинические проявления, по сути, повторяют симптомы оптикомиелита: оптиконеврит, LETM, поражение циркумвентрикулярных органов и аутоиммунная AQP4-миопатия (табл. 2). Оптикомиелит и NMOSD могут развиваться при онкологических заболеваниях (4–5 % от всех случаев NMOSD и 15 % у пациентов старшего возраста) в форме паранеопластического синдрома. Тем не менее наличие только AQP4-IgG без клинических проявлений поражения ЦНС не позволяет поставить диагноз NMOSD.
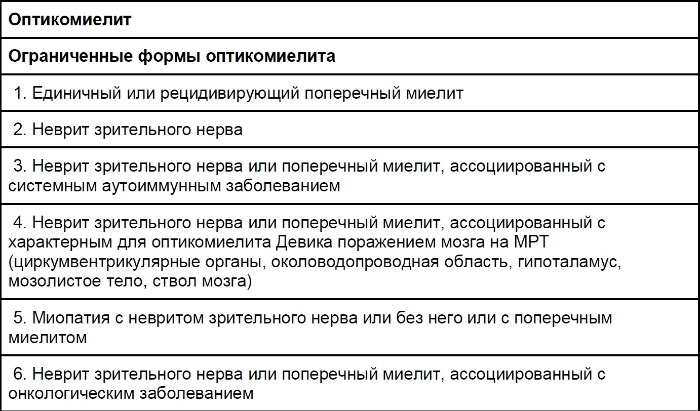 Таблица 2 ❘ Спектр оптикомиелит-ассоциированных расстройств (NMOSD), определяемых как AQP4-IgG-серопозитивный статус (данные 2015 года).
Таблица 2 ❘ Спектр оптикомиелит-ассоциированных расстройств (NMOSD), определяемых как AQP4-IgG-серопозитивный статус (данные 2015 года).
Поражение головного мозга
Вовлечение в патологический процесс головного мозга, по данным МРТ, происходит в 60 % случаев при первом приступе, при этом в 10 % случаев патология может напоминать таковую при РС. Еще 10 % пациентов имеют типичное для оптикомиелита расположение очагов поражения (гипоталамус, перивентрикулярные области) (рис. 3). Заболевание может протекать бессимптомно или иметь клинику поражения циркумвентрикулярных органов: неукротимая тошнота и рвота (area postrema и дно четвертого желудочка), икота, синдром неадекватной секреции антидиуретического гормона, нарколепсия или анорексия (гипоталамус) или эндокринопатии заднего отдела гипофиза. Ствол мозга поражается в 31 % случаев (чаще у небелых пациентов), что проявляется рвотой (33 %), икотой (22 %), глазодвигательными нарушениями (20 %) и зудом (12 %), в 3,3 % может произойти потеря слуха, паралич лицевого нерва, вестибулопатия, невралгия тройничного нерва. Сообщалось также о 5 женщинах с синдромом задней обратимой энцефалопатии. Клиническая манифестация и поражения головного мозга у детей могут напоминать острый рассеянный энцефаломиелит (ОРЭМ).
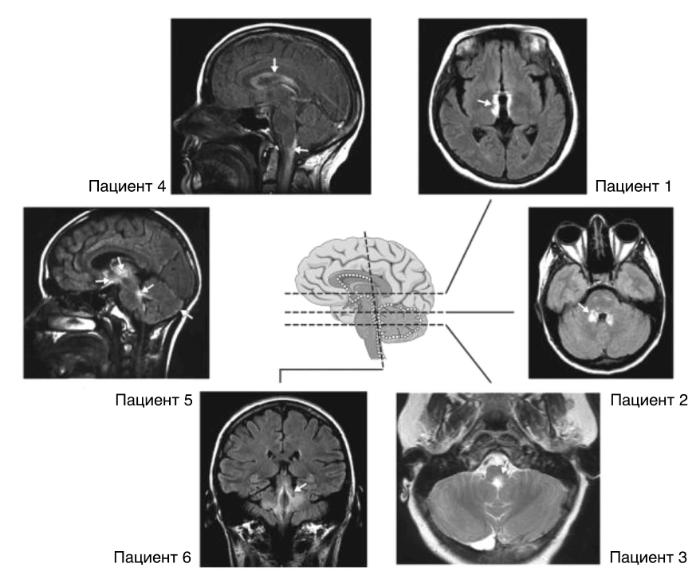
Пациент 1: локализация вокруг 3-го желудочка и гипоталамуса; Пациент 2: локализация вокруг 4-го желудочка; Пациент 3: локализация вокруг 4-го желудочка и водопровода; Пациент 4: периэпендимальнаяя локализация, поражения вокруг боковых желудочков, очаг поражения спинного мозга с распространением на ствол мозга; Пациент 5: поражение таламуса, гипоталамуса, хиазмы, а также поражение вокруг 4-го желудочка, субпиальное поражение в полушариях мозжечка; Пациент 6: локализация вокруг 4-го желудочка и ножек мозжечка.
Сопутствующие аутоиммунные заболевания
Органоспецифические и неспецифические аутоиммунные проявления (аутоиммунные заболевания или антительные маркеры) часто сопутствуют NMOSD. Например, синдром Шегрена и системная красная волчанка. Поперечный миелит всегда является проявлением NMOSD, а не осложнением васкулита при системных заболеваниях соединительной ткани. Аутоиммунная miastenia gravis встречается в 100 раз чаще у пациентов с NMOSD (2 %), чем в целом в популяции (0,02 %). Также отмечены случаи развития целиакии, аутоиммунного гастрита, аутоиммунного тиреоидита, язвенного колита и склерозирующего холангита.
Поражение скелетной мускулатуры
Гиперкалиемия может предшествовать или сопутствовать NMOSD. Показателен случай с LETM и оптикомиелитом, когда в ответ на терапию кортикостероидами появились симптомы проксимальной миалгии (КФК >10000 ЕД/л). Биопсия пораженной мышцы показала потерю сарколеммами AQP4, а также линейные отложения IgG и белков системы комплемента, в мышце наблюдалась характерная для поражения ЦНС гистологическая картина: лимфогистиоцитарные инфильтраты и некоторое количество эозинофилов. Здоровые мышцы имеют на своей поверхности большое количество каналов AQP4, а при некротизирующих аутоиммунных миопатиях и дерматомиозитах также происходит частичная потеря AQP4, но без отложения иммунных комплексов в сарколемме.
Миелин-олигодендроглиоцитарный гликопротеин (MOG)
Антитела к миелин-олигодендроглиоцитарному гликопротеину (MOG-IgG) ранее считались маркерными антителами при детском РС, сегодня они обнаруживаются у детей с ОРЭМ, а также иногда у взрослых и детей в случае рецидива AQP4-IgG-позитивного и AQP4-IgG-негативного оптикомиелита. MOG-IgG-позитивные случаи чаще всего монофазны, мужчины болеют чаще, возраст манифестации меньше, на МРТ чаще отмечаются поражения глубокого серого вещества, а прогноз заболевания более благоприятный. Следует отметить, что иммуногистохимическими методами данные случаи не исследовались.
Пациенты с установленными диагнозами оптикомиелита (MOG-IgG-позитивными и AQP4-IgG-негативными фенотипами) имели в ликворе высокий уровень основного белка миелина при отсутствии глиального фибриллярного кислого протеина (GFAP), что говорит в пользу демиелинизирующего процесса, а не астроцитопатии.
Пока не будут получены морфологические данные в отношении MOG-IgG-позитивных случаев, корректнее называть их «MOG-аутоиммунный неврит зрительного нерва, поперечный миелит или энцефаломиелит», а не NMOSD.
Лабораторно-инструментальные исследования
МР-особенности
Нейровизуализация поперечного миелита обычно выявляет поражения спинного мозга протяженностью более 3 позвонков, однако в 14 % случаев в начале заболевания область поражения может быть короче. Протяженные очаги отечны, неравномерно накапливают контраст, иногда отмечаются участки центрального некроза или полости. Позднее можно наблюдать фокальную атрофию спинного мозга. Поражение центрального серого вещества помогает отличить данную патологию от демиелинизирующих заболеваний. ДТИ (диффузионно-тензорные изображения) выявляют у пациентов в течение года после последнего обострения более выраженную, чем при РС, радиальную диффузию в белом веществе, что соответствует большей деструкции тканей.
К особенностям неврита зрительного нерва при оптикомиелите можно отнести билатеральный и протяженный характер поражения, поражение хиазмы и зрительных трактов. Оптическая когерентная томография и зрительные вызванные потенциалы дополняют и завершают инструментальную диагностику. В головном мозге наиболее характерны поражения гипоталамуса и перивентрикулярной области, но в 10 % случаев можно наблюдать РС-подобную картину, которая отвечает критериям Баркофа. [1]
Были исследованы характерные особенности визуализации NMOSD и РС (табл. 3,4,5). Два опытных нейрорадиолога, у которых отсутствовали данные клинического осмотра и диагноза, оценили каждое поражение на МРТ. Это исследование показало, что распределение повреждений значительно различается между NMOSD и РС. Хотя было обнаружено, что округлые поражения/пальцы Доусона и изолированное поражение U-волокон/юкстакортикального белого вещства имели значительное более высокую частоту при РС, нежели при NMOSD, по сравнению с предыдущими европейскими исследованиями, частота данного типа поражений при NMOSD в этом исследовании (округлые поражения 21,5 %, изолированное поражение U-волокон 10,1 %) была выше. Прошлые исследования показали, что наличие таких поражений может развести РС и NMOSD с высокой чувствительностью и специфичностью, хотя это спорно для азиатской популяции с NMOSD. Сообщалось, что генетические факторы или факторы окружающей среды влияют на различия в визуализации у пациентов с РС в разных областях. Визуализационные особенности при NMOSD также могут иметь различия у пациентов из Азии и Европы.
Несмотря на то, что облаковидное усиление контрастирования является характерным паттерном контрастирования для NMOSD, ни у одного пациента с NMOSD в этом исследовании не было обнаружено этого признака. Облаковидное усиление контрастирования определяется как «множественное пятнистое усиление с размытыми краями в смежных областях по сравнению с изолированными усилением очага поражения». Однако диагностический порог для этого признака может немного отличаться в зависимости от оценщика, что может повлиять на результаты исследования.
Хотя двусторонний неврит зрительного нерва и поражение хиазмы являются специфическими особенностями NMOSD, в данном исследовании не было обнаружено существенных различий между NMOSD и РС. Результаты могут быть объяснены следующим: во-первых, пациенты подбирались как в острой, так и в хронической фазах, тогда как в предыдущие исследования были включены только пациенты в острой фазе; во-вторых, в этот ретроспективный анализ поражений зрительного нерва было включено различное количество пациентов с NMOSD и РС. Таким образом, требуются дальнейшие проспективные исследования с четкими критериями соответствия. [2]
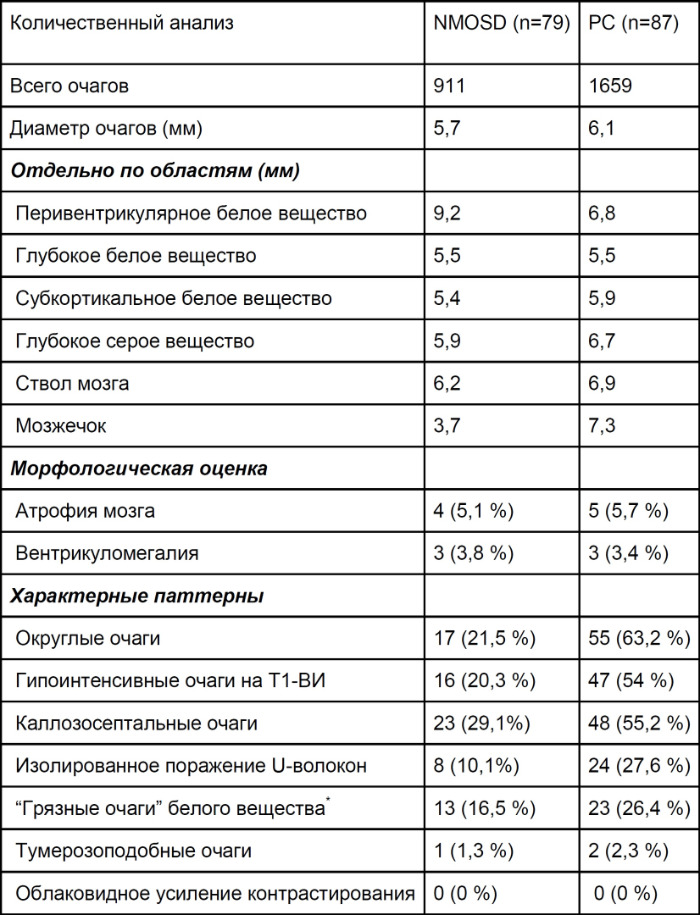 Таблица 3 ❘ Характеристика очагов поражения головного мозга при РС и NMOSD. Приведены медианные значения. [2]
* грязные очаги определяются, как очаги с промежуточной интенсивностью сигнала между сигналами от очаговых поражений и нормально выглядящим белым веществом
Таблица 3 ❘ Характеристика очагов поражения головного мозга при РС и NMOSD. Приведены медианные значения. [2]
* грязные очаги определяются, как очаги с промежуточной интенсивностью сигнала между сигналами от очаговых поражений и нормально выглядящим белым веществом
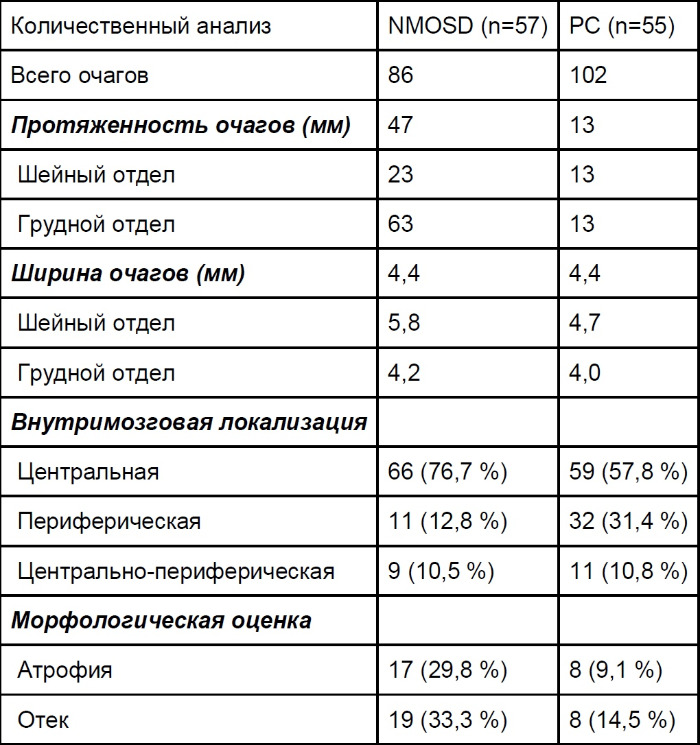 Таблица 4 ❘ Характеристика очагов поражения спинного мозга при РС и NMOSD. Приведены медианные значения. [2]
Таблица 4 ❘ Характеристика очагов поражения спинного мозга при РС и NMOSD. Приведены медианные значения. [2]
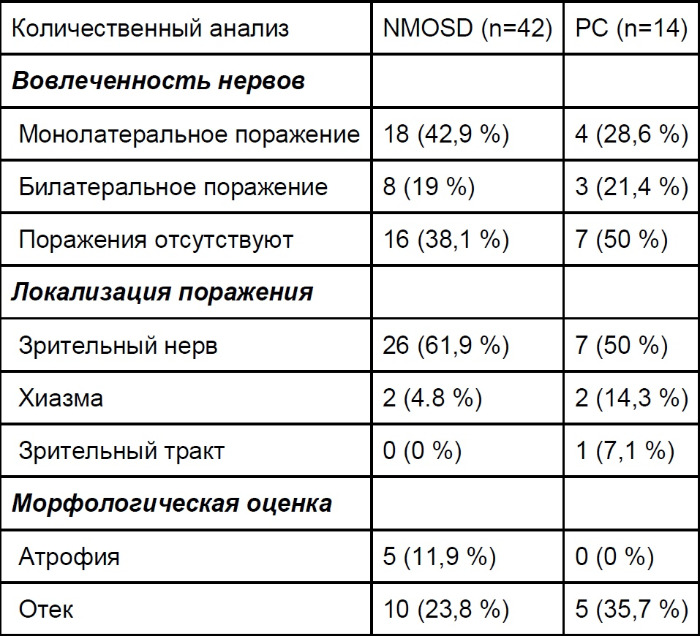 Таблица 5 ❘ Характеристика очагов поражения зрительного нерва при РС и NMOSD. Приведены медианные значения. [2]
Таблица 5 ❘ Характеристика очагов поражения зрительного нерва при РС и NMOSD. Приведены медианные значения. [2]
Изменения ликвора
Изменения ликвора при оптикомиелите довольно сильно отличаются от таковых при РС. Только 16 % AQP4-IgG-позитивных оптикомиелитов имеют олигоклональные иммуноглобулины, у 50 % наблюдается плейоцитоз: в среднем около 19/мкл лейкоцитов, причем у 6,2 % он достигает 100/мкл, в основном за счет нейтрофилов, базофилов и эозинофилов.
Уровень GFAP, маркера повреждения астроцитов, более высокий при оптикомиелите, чем при РС и других неврологических заболеваниях, в то же время цитоплазматический белок S100B, который содержится преимущественно в астроцитах, повышается умеренно. Основной белок миелина и тяжелые цепи нейрофиламентов показывали сходные уровни сразу для трех заболеваний. Концентрация GFAP нормализуется практически сразу после начала терапии, в то же время уровень основного белка миелина остается высоким, вероятно, маркируя демиелинизацию. Высокие уровни основного белка миелина, S100B и GFAP при обострении коррелируют с протяженностью LETM и ухудшают показатели EDSS (Expanded Disability Status Scale — расширенная шкала оценки степени инвалидизации). Значения данной шкалы спустя 6 месяцев коррелируют только с уровнем GFAP при обострении.
Антительный профиль
Огромное количество аутоантител (органспецифические и неспецифические) помогает различить оптикомиелит и РС, при этом титры в целом невысокие (табл. 6). Сообщалось об обнаружении антител к каналам аквапорина-1, но их клиническая польза пока не оценивалась.
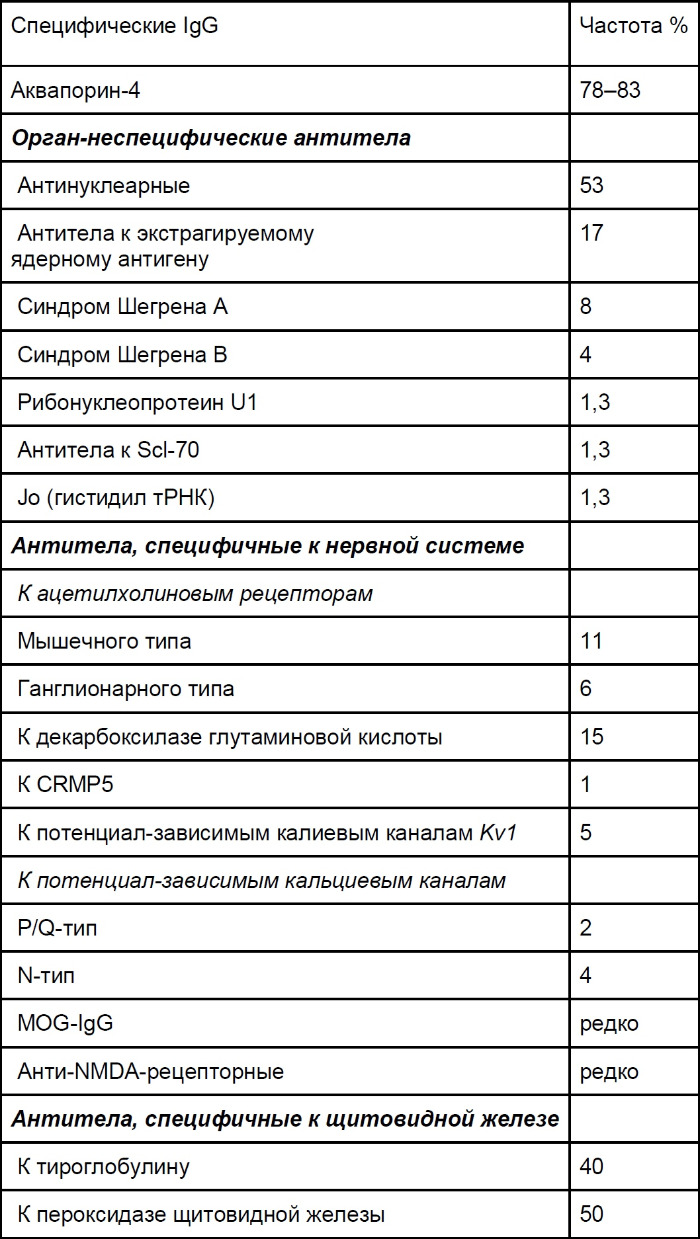 Таблица 6 ❘ Антительный профиль оптикомиелита [1]
Таблица 6 ❘ Антительный профиль оптикомиелита [1]
Прогноз
До открытия AQP4-IgG прогноз оптикомиелита был крайне неблагоприятен, т. к. терапевтические возможности были не определены. В обзоре серии случаев Wingerchuk et al. 1999 года сообщалось, что при манифестации оптикомиелита развитие основных симптомов происходит быстро, в среднем, в течение 5 дней, с последующим умеренным восстановлением. Впоследствии (в случае рецидива) интервал между первоначальным LETM и невритом зрительного нерва увеличивается в среднем до 166 дней, тяжелые повторные приступы отмечаются в течении 3 лет, а инвалидизация происходит ступенчато. Треть пациентов умирает от респираторных осложнений.
По результатам исследования AQP4-IgG-позитивных пациентов из Великобритании и Японии 2012 года, постоянное двустороннее нарушение зрения развивается у 18 % пациентов (средняя продолжительность заболевания у них составляет 75 месяцев), постоянные двигательные нарушения (невозможность пройти более 100 метров без помощи) — у 34 %, прикованы к инвалидному креслу 23 %. При средней продолжительности заболевания около 99 месяцев 9 % пациентов умирает после 4,5 приступов. В английском когортном исследовании у афро-карибских пациентов отмечались более ранний возраст манифестации болезни и более высокая вероятность развития зрительных нарушений, чем у белых пациентов.
По данным отчета 2013 года, в котором рассматривались AQP4-IgG-позитивные и AQP4-IgG-негативные пациенты, 65 % больных теряли зрение в среднем через 8,3 года после манифестации болезни (50 % монолатерально, 15 % билатерально), и более трети не могли передвигаться без посторонней помощи.
Данные говорят о более благоприятном прогнозе заболевания после открытия AQP4-IgG, предположительно, из-за возможности ранней диагностики и, как следствие, раннего начала лечения, направленного на снижение уровня иммуноглобулинов, блокирование активации системы комплемента и действия интерлейкина-6.
Лечение
Терапия оптикомиелита преследует две цели: купирование острого состояния (с целью минимизировать неврологический дефицит и повысить шанс на восстановление) и предотвращение рецидивов, приводящих к инвалидизации. Терапевтические решения должны приниматься индивидуально на основании клинической картины и не должны опираться на уровень AQP4-IgG. На сегодняшний день проспективные контролируемые исследования, оценивающие варианты терапии оптикомиелита (табл. 7), отсутствуют.
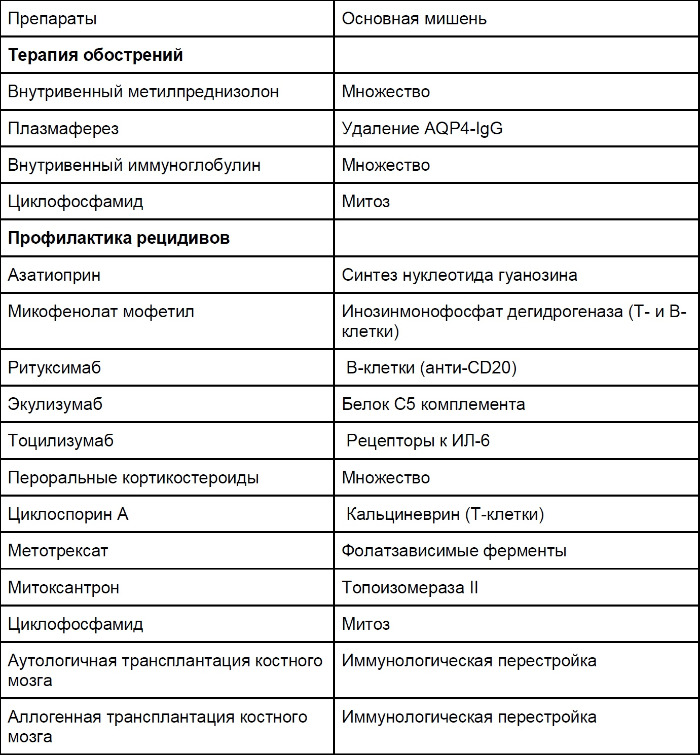 Таблица 7 ❘ Терапия оптикомиелита [1]
Таблица 7 ❘ Терапия оптикомиелита [1]
Как и для других IgG-опосредованных аутоиммунных заболеваний, внутривенный метилпреднизолон (ВВМПД) и плазмаферез считаются оптимальными вариантами терапии первой линии. Рекомендовано применение 1 гр ВВМПД от 3 до 5 дней. Пероральные формы преднизолона используются в качестве профилактики раннего рецидива с последующим переходом на нестероидные иммуносупрессоры. При тяжелых обострениях одного ВВМПД может оказаться недостаточно, добавление к терапии плазмафереза (от 5 до 7 сеансов) улучшает результаты терапии. В некоторых особо тяжелых случаях циклофосфамид (750–1000 мг/м2) оказывался эффективнее пульс-терапии метилпреднизолоном.
Внутривенные иммуноглобулины (ВВИГ) совместно с плазмаферезом или без него показали хороший результат в исследовании на 10 пациентах, которые не получали ВВМПД.
Для определения оптимальных терапевтических возможностей, учитывающих возраст пациентов, сопутствующие заболеваний и тяжесть приступов необходимы дальнейшие исследования.
Профилактика рецидивов
Иммуномодулирующая терапия, применяемая для РС, в случае NMOSD не эффективна, а некоторые препараты могут вызвать ухудшение. Ретроспективно, используя доступные данные большого количества пациентов, свою эффективность показал ряд препаратов: азатиоприн, микофенолат мофетил и ритуксимаб.
Азатиоприн (не менее 2 мг/кг/сутки) снизил частоту ежегодных рецидивов в исследовании на 99 пациентах. Показатели крови, функции печени и почек должны контролироваться на всем протяжении лечения, а также желательно определить активность тиопурин метилтрансферазы (фермент, ответственный за активацию азатиоприна) перед началом лечения. Поскольку эффект от азатиоприна отсроченный, в первые несколько месяцев применяют пероральные формы глюкокортикостероидов. В исследовании 32 пациентов азатиоприн снизил частоту рецидивов на 72 %, однако был неэффективен у половины пациентов, несмотря на комбинацию с преднизолоном.
Микофенолат мофетил также может использоваться для длительной иммуносупрессии. В исследовании 28 пациентов частота рецидивов снизилась на 87 %.
Ритуксимаб, анти-CD20 моноклональный препарат, угнетающий B-клеточное звено, показал высокую эффективность в терапии NMOSD. Открытые исследования с использованием различных протоколов дозирования и мониторинга CD19+ лимфоцитов предполагают, что применение препарата следует возобновлять, когда в крови снова появляются В-клетки. Частота ежегодных рецидивов уменьшается до 87 %, а 60–70 % пациентов не имеют рецидивов в течении следующих 3 лет.
Экулизумаб, моноклональный препарат, который связывается с белком С5 комплемента, показал отсутствие рецидивов в течении года у 12 из 14 пациентов в пилотном открытом исследовании. Во время лечения медиана частоты рецидивов была снижена с 3 до 0, а показатель EDSS улучшился с 4,3 до 3,5.
Тоцилизумаб, моноклональный препарат против рецепторов ИЛ-6, показал эффективность в открытом исследовании у 7 пациентов. Через год частота рецидивов снизилась с 2,96 до 0,4. Показатели EDSS были значительно снижены, нейропатическая боль и общая слабость отмечались пациентами реже.
Пероральные кортикостероиды в качестве монотерапии или дополнительной терапии предотвращают рецидивы, что было показано в небольшом ретроспективном исследовании. Комбинированная терапия сводит к минимуму побочные явления расширенной терапии кортикостероидами. При длительном лечении рекомендуется проводить профилактику остеопороза и мониторинг глюкозы крови.
Циклоспорин А применяют в комбинации с пероральными кортикостероидами. Ежегодная частота рецидивов у 9 пациентов, получавших лечение, снизилась с 2,7 до 0,38.
В ретроспективном исследовании 14 пациентов метотрексат показал значительное снижение частоты рецидивов (0,18 во время терапии и 1,39 до), 43 % пациентов не имели рецидивов, а скорость инвалидизации стабилизировалась или уменьшилась на 79 %.
Применение циклофосфамида позволило снизить медианный показатель EDSS с 8 до 5,75 у 4 пациентов с LETM.
Аутологичная трансплантация костного мозга остановила прогрессирование заболевания у 3 из 16 пациентов, у которых отмечалась устойчивость к проводимому ранее лечению. Эти пациенты прекратили получать какую-либо терапию (медиана 47 месяцев). Остальные 13 пациентов нуждались в продолжении терапии из-за ухудшения общих показателей или рецидивов. Аллогенная трансплантация костного мозга была эффективна у 2 пациентов с агрессивным оптикомиелитом, у которых проводимая ранее терапия, включая аутологичную трансплантацию костного мозга, не дала эффекта. Оба пациента не имели рецидивов в течении 36 и 48 месяцев.
Патогенез
Благоприятный эффект от применения плазмафереза и препаратов, подавляющих активность В-лимфоцитов, предполагает, что основную роль в патогенезе играют AQP4-IgG.
Иммунопатогенез
Вокруг гиалинизированных сосудов выявляются продукты активации комплемента, которые связываются с молекулами IgG. Преобладание полиморфно-ядерных провоспалительных клеток согласуется с участием IgG и комплемента в патогенезе оптикомиелита. Потеря каналов аквапорина-4 астроцитами, выжившими в сублитических поражениях ЦНС, отличает оптикомиелит от РС и может свидетельствовать о потенциально обратимых гистопатологических изменениях. В некоторых очагах поражения наблюдается выраженное воспаление и потеря астроцитами AQP4, но при этом сохраняется GFAP. Другие очаги демонстрируют невысокую астроцитарную иммунореактивность в отношении GFAP при относительно сохранном миелине. Эти данные говорят в пользу того, что утрата AQP4 является причиной поражения астроцитов, что в итоге приводит к вторичной демиелинизации. В дальнейшем в сером и белом веществе спинного мозга отмечается обширная демиелинизация, кавитация, некроз и утрата аксонов (сфероидоз).
Роль AQP4-IgG
Появление IgG в ответ на белковые аутоантигены отражает взаимодействие регуляторных и хелперных Т-лимфоцитов с антиген-специфическими В-лимфоцитами. Роль AQP4-специфичных Т-лимфоцитов неясна, однако они могут способствовать повреждению гематоэнцефалического барьера. В изученных очагах поражения оптикомиелита обнаруживаются IgG-продуцирующие плазматические клетки. Появляется все больше сведений об интратекальном синтезе AQP4-IgG, высокий уровень которого в спинномозговой жидкости достоверно коррелирует с тяжестью обострения. В плазме крови выявляется повышенный уровень ИЛ-6, который способствует выживанию лимфоцитов.
Для запуска патологического процесса требуется связывание IgG с внеклеточным доменом AQP4. AQP4-IgG являются преимущественно Ig подкласса G1 — потенциальным активатором системы комплемента. В мембране астроцитов AQP4 существует в виде тетрамера и имеет две изоформы: полную (М1) и короткую (М23).
M23-AQP4 спонтанно образует ортогональные массивы частиц, а большинство гомомерных М1 тетрамеров остаются синглетными.
При воздействии AQP4-IgG астроциты быстро интернализуют тетрамеры М1, в то же время связывание М23 вызывает их слияние в более крупные массивы, которые превышают способность астроцитов к эндоцитозу. Когда комплемент становится доступным, крупные массивы AQP4, в которых находятся плотно упакованные Fc-хвосты IgG1, лавиноообразно активируют систему комплемента, способствуя привлечению лейкоцитов, повышению сосудистой проницаемости и массивному пропотеванию плазмы, богатой IgG, белками системы комплемента, которые при связывании с комплексом антиген-антитело будут способствовать усилению активации комплемента.
По данным исследований in vitro, выявлена взаимосвязь тяжести атаки оптикомиелита и степени опосредованного комплементом повреждения клеток, экспрессирующих AQP4, на фоне воздействия IgG у пациентов с оптикомиелитом (6 пациентов с тяжелым обострением, 6 пациентов с легким, p=0,05). Дегрануляция NK-клеток, которые активируются по комплемент-независимому пути посредством связывания IgG с AQP4 (антителозависимая клеточная цитотоксичность), также вызывает гибель астроцитов in vitro.
Блокирование антителами тока воды вполне правдоподобно объясняет видимый отек миелина в ранних очагах оптикомиелита, который является возможным предшественником демиелинизации. Антитела, потенциально способные блокировать ток воды, независимо от поражения AQP4 (т. е. температурно независимые), продемонстрировали данный эффект у шпорцевых лягушек (Xenopus). Данные антитела были получены у по меньшей мере 50 пациентов с оптикомиелитом и высоким уровнем содержания AQP4-IgG. Таким образом, были получены данные, оспаривающие основную роль AQP4-IgG, хотя стоит учитывать, что они были основаны на исследовании моноклональных AQP4-IgG и сывороток от относительно небольшого количества отдельных пациентов с помощью технологии квантовых точек.
Еще одно свойство комплекса AQP4-IgG — нарушение гомеостаза глутамата. Транспортер возбуждающих аминокислот EAAT2 нековалентно связан с аквапориновыми каналами на пресинаптической мембране астроцитов. На EAAT2 приходится до 90 % утилизированного глутамата в ЦНС, кроме того, данный транспортер может поглощаться AQP4-IgG. Применение данных антител на культуре клеток астроцитов приводило к снижению функционального поглощения глутамата. Более того, в подострых очагах поражения в спинном мозге в центральном сером веществе наблюдается выраженный дефицит EAAT2. Таким образом, особая уязвимость олигодендроцитов к токсичности глутамата является еще одним вероятным объяснением демиелинизации при оптикомиелите.
Животные модели
Животные модели, описанные на сегодняшний день, воспроизводят некоторые гистопатологические характеристики оптикомиелита, но не клинические данные. Поражения, свойственные оптикомиелиту, не возникают, когда сывороточный IgG от больных пациентов вводят крысам с интактным гематоэнцефалическим барьером. Однако, когда его применяют у крыс с экспериментальным аутоиммунным энцефалитом, у них развивается весь спектр ожидаемых патофизиологических изменений. Также IgG не вызывали никаких изменений у молодых крыс, у которых содержание эндотелиальных, т. е. барьерных, клеток в ЦНС крайне мало.
С другой стороны, сообщалось о повреждении астроцитов и потере AQP4 в спинном мозге после введения IgG мышам, предварительно получавших полный адъювант Фрейнда в комбинации с коклюшным токсином или без него. Авторы предположили, что микобактериальный адъювант способствует повышению проницаемости гематоэнцефалического барьера. Очаговые поражения, характерные для оптикомиелита, развивались у мышей, которым вводился IgG непосредственно в полушария головного мозга, но только в том случае, если также вводили человеческую плазму в качестве источника комплемента. [1]
Источники
1. Zekeridou A., Lennon V. A. Aquaporin-4 autoimmunity //Neurology-Neuroimmunology Neuroinflammation. – 2015.
2. Tatekawa H. et al. Imaging Differences between Neuromyelitis Optica Spectrum Disorders and Multiple Sclerosis: A Multi-Institutional Study in Japan //American Journal of Neuroradiology. – 2018.
3. Chee C. G. et al. MRI Features of Aquaporin-4 Antibody–Positive Longitudinally Extensive Transverse Myelitis: Insights into the Diagnosis of Neuromyelitis Optica Spectrum Disorders //American Journal of Neuroradiology. – 2018.
