
Эстроген-индуцированный апоптоз
Апоптоз — один из важнейших типов регулируемой гибели клеток в процессе онтогенеза и поддержании гомеостаза нормальных зрелых тканей. Митохондриальный (внутренний) путь апоптоза контролируется соотношением анти- и проапоптотических факторов из семейства белков B-клеточных лимфом 2 (Вcl-2). Молекулярные механизмы регулирования экспрессии данных белков крайне многообразны и представлены различными внешними и внутренними стимулами, в том числе действием биологически активных веществ.
В процессе эмбрионального развития происходит не только бурный рост и пролиферация, но и обновление клеток, часть которых самоотверженно погибает для более эффективного онтогенеза. Апоптоз начинает играть важную роль уже на стадии имплантации, при этом его активность строго контролируется. На стадии приближения и первичного контактирования бластоцисты с эндометрием секретируются факторы, ингибирующие вступление клеток эндометрия в апоптоз. Далее, в фазе адгезии, стимулируется апоптоз в клетках эндометрия вокруг прикрепленной бластоцисты, что облегчает ее инвазию. Также структуры трофобласта индуцируют апоптоз в эндотелиоцитах и гладкомышечных клетках матки, что приводит к ремоделированию спиральных артерий для обеспечения оптимального кровоснабжения растущего плода [1].
В период гестации важную роль играет гормональная регуляция, осуществляемая всеми компонентами эндокринной системы, и особенно половыми гормонами. Зачастую на первый план выходит прогестерон, получивший титул «гормона беременности», но вклад эстрогенов также значителен. Их воздействие на морфогенез и функциональные свойства эмбриональных тканей зависит от срока гестации и концентрации гормона. Обнаружено, что высокий уровень эстрадиола (E2) отрицательно влияет на имплантацию бластоцисты. Существует также повышенный риск преждевременных родов и рождения ребенка с низкой массой тела у женщин с высоким уровнем E2 в сыворотке крови в первом триместре беременности. Высокий Е2 способствует субоптимальной перфузии развивающейся плаценты и снижению кровотока в маточной артерии, что связывают с его проапоптотическими и антипролиферативными эффектами [2].
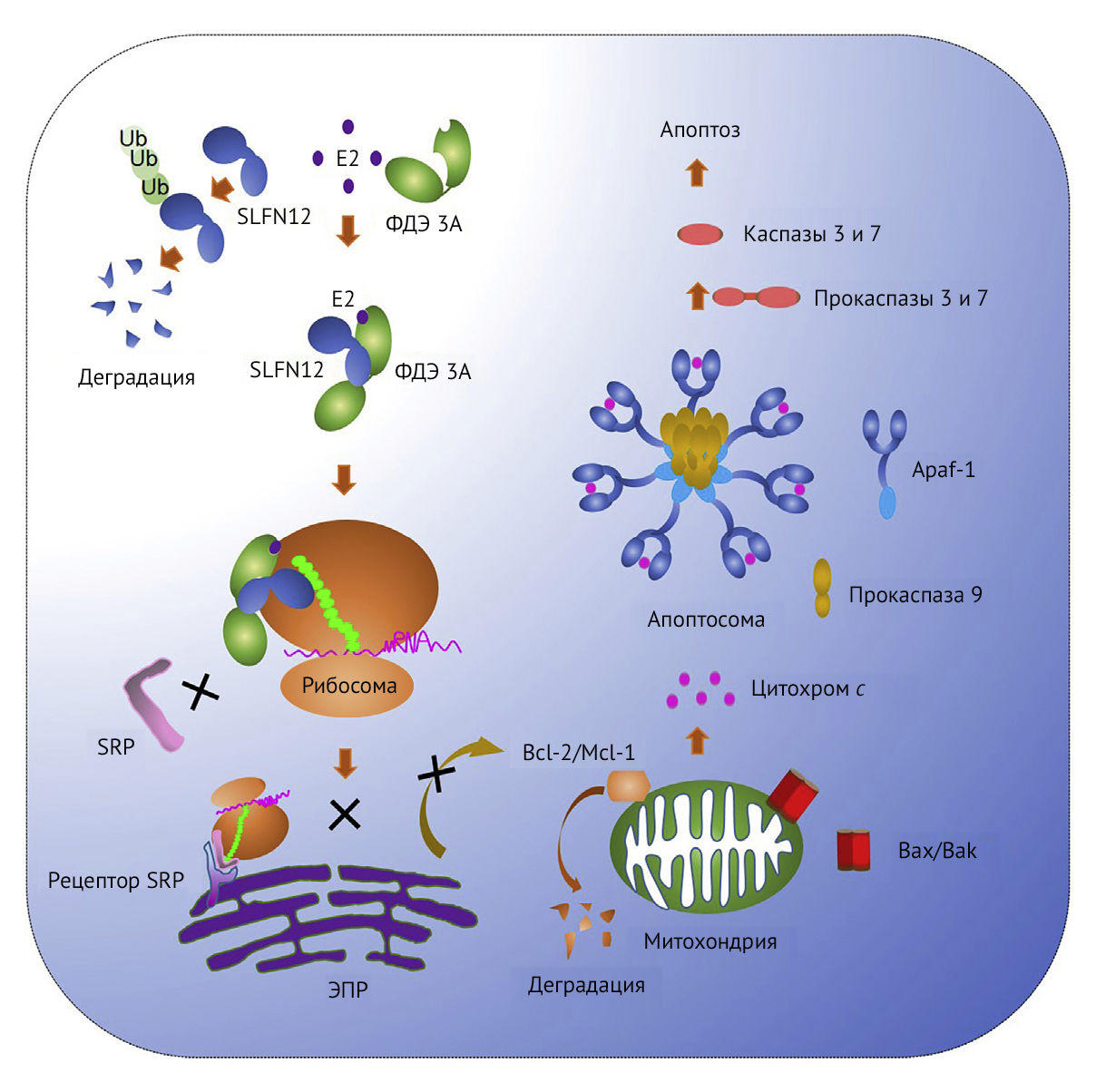
Запуск программы клеточной смерти обусловлен эстроген-зависимым подавлением экспрессии белков Bcl-2 и Mcl-1 (дифференцировочный белок клеток миелоидного лейкоза). При этом вместо классических рецепторов стероидных гормонов молекулы внеклеточного Е2 связываются с ферментативно активным доменом фосфодиэстеразы 3A (ФДЭ 3А). Это небольшая молекула с рецепторной активностью, которая обладает тропностью к разнообразным соединениям (например, циклическому аденозинмонофосфату, циклическому гуанозинмонофосфату и E2). Кроме того, ФДЭ 3А способна образовывать связь с молекулой 6-(4-(диэтиламино)-3-нитрофенил)-5-метил-4,5дигидропиридазин-3(2Н)-она (DNMDP), являющейся цитотоксическим пиридазином [3]. Связь рецептор-лиганд вызывает конформационные изменения белка семейства Schlafen (SLFN12), что стабилизирует его молекулу. В обычных условиях концентрация SLFN12 в клетках крайне низка из-за его убиквитин-опосредованной деградации. Субстрат образовавшегося комплекса Е2-ФДЭ 3А-SLFN12 — рибосома, где SLF12 связывается с рибосомальной РНК, блокируя трансляцию белка. Также дополнительно он ингибирует связывание частиц распознавания сигналов (SRP) со специфической сигнальной последовательностью синтезируемого белка, что нарушает трансляцию [4].
В результате снижается экспрессия антиапоптотических мембранных белков Bcl-2 и Mcl-1, что запускает активность проапоптотических белков Bax и Bak. Их олигомеризация приводит к образованию пор в наружной мембране митохондрий, через которые цитохром С из митохондриального межмембранного пространства выходит в цитоплазму. Далее активируются каспазы 9 и 3. Так происходит гибель клетки с помощью внутреннего пути апоптоза (рис. 1) [5].
.
Белок SLFN12 и маркеры активации апоптоза обнаружены в клетках синцитиотрофобласта на фоне высокого уровня эстрогеновых гормонов [5].
Уровень эстрадиола, достаточно высокий для запуска апоптоза, отмечается в ворсинчатом трофобласте. Он состоит из многочисленных структур цитотрофобласта, которые образуют многоядерный синцитиотрофобласт посредством слияния клеток [5].
Интенсивность процессов апоптоза планомерно увеличивается в плацентарной ткани до родоразрешения. Эстроген-контролируемые механизмы в норме обеспечивают динамическое обновление клеток за счет апоптоза, что является критическим для успешной имплантации яйцеклетки и дальнейшего развития плаценты. Гиперпродукция активного эстрадиола, особенно на ранних сроках гестации, нарушает этот баланс. Специфичным маркером эстроген-индуцированного механизма апоптоза может стать белок SLFN12, уровень которого значительно повышается в ходе сигнального каскада запрограммированной клеточной гибели.
Источники:
- Stenhouse C. et al. Associations between fetal size, sex and both proliferation and apoptosis at the porcine feto-maternal interface.Placenta.2018;70:15-24.
- Patel S., Kilburn B., Imudia A., Armant D.R., Skafar D.F. Estradiol elicits proapoptotic and antiproliferative effects in human trophoblast cells. Biology of reproduction. 2015;93(3):74-1.
- Lewis-Wambi J., Jordan V. Estrogen regulation of apoptosis: How can one hormone stimulate and inhibit?. Breast cancer research.2009;11:206.
- McDaniel R.E. et al. Estrogen-mediated mechanisms to control the growth and apoptosis of breast cancer cells: a translational research success story. Vitam Horm. 2013;93:1-49.
- Li D. et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Molecular Cell. 2019;75:1–14.
