
Роль ионных каналов в патогенезе сахарного диабета
ATФ-чувствительные калиевые (К+-АТФ) каналы, расположенные в β-клетках поджелудочной железы, играют важную роль в метаболизме глюкозы и секреции инсулина. Они контролируют возбудимость клеточной мембраны, поддерживая тем самым уровень глюкозы в крови в пределах физиологического диапазона. Нарастание уровня глюкозы приводит к повышению внутриклеточного соотношения ATФ/АДФ и закрытию калиевых каналов. В ответ происходит деполяризация мембраны. Открываются потенциал-зависимые кальциевые каналы и увеличивается концентрация ионов кальция [Ca2+], что в свою очередь стимулирует секрецию инсулина. И наоборот: снижение уровня глюкозы открывает калиевые АТФ-чувствительные каналы, что подавляет электрохимический триггер секреции инсулина [1].
В условиях длительной гипергликемии закономерно ожидать компенсаторного увеличения массы β-клеток, что обеспечит увеличившуюся потребность в инсулиносекреции. В случае неспособности β-клеточного пула поддерживать необходимый уровень секреции инсулина и развивается сахарный диабет.
Но при стабильно высокой гипергликемии дисфункция β-клеток неуклонно нарастает. Это так называемый феномен глюкозотоксичности [2].
Одним из ее механизмов является уникальная способность β-клеток повышать активность окислительного фосфорилирования в ответ на повышение уровня глюкозы в крови. Агрессивным побочным продуктом этой реакции являются активные формы кислорода, нарушающие работу митохондрий. Нарастают маркеры окислительного стресса, что в итоге приводит к апоптозу [3].
Однако эта парадигма гибели β-клеток в настоящее время подвергается сомнению из-за обнаружения потери идентичности β-клеток поджелудочной железы и их дедифференцировки при сахарном диабете [4].
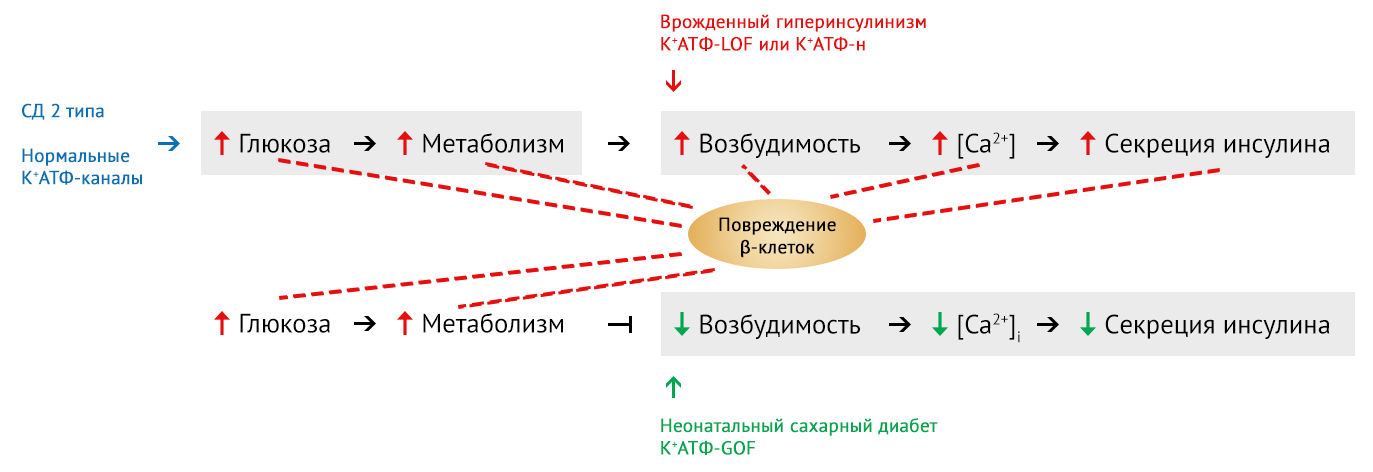
Тем не менее негативные эффекты аномально высокого или низкого уровня ионов кальция порождают вопрос: какова роль мембранной гипер- или гиповозбудимости в функционировании островков?
В ходе эксперимента, заключающегося в инкубации клеточных линий островков мышей с генетически повышенной (мышиная линия с нокаутом калиевых ATФ-каналов К+АТФ-н) или пониженной мембранной возбудимостью (мышиная линия с мутацией усиления функции (gain-of-function) К+ATФ-GOF) при стабильно высоком уровне глюкозы in vitro, была осуществлена попытка отделить роль мембранной возбудимости от «примеси» других факторов, неизменно наблюдающихся in vivo (в живом организме) при любой реакции на глюкозотоксичность.
Согласно данным in vitro, повышение возбудимости клеточных мембран, достигнутое либо генетически (нокаут K+ATФ-н), либо фармакологически (островки, обработанные глибенкламидом), действительно приводит к заметному снижению уровня инсулина в течение примерно 3 дней. И наоборот: в островках с генетически (K+ATФ-GOF) или фармакологически (обработанные активатором калиевых каналов диазоксидом) сниженной возбудимостью клеточной мембраны поддерживается необходимое содержание инсулина в течение полных 10 дней даже в условиях инкубации в среде с высоким содержанием глюкозы [5].
При этом долгосрочные результаты in vivo у мышей с измененной возбудимостью мембраны оказываются противоречивыми. Отмечено значительное снижение содержания инсулина и его мРНК, а также уменьшение массы β-клеток в островках мышей с диабетом К+ATФ-GOF со сниженной возбудимостью мембран. Это является результатом длительного течения сахарного диабета [5].
Напротив, in vivo в островках с искусственно повышенной возбудимостью клеточной мембраны и повышенным уровнем ионов кальция (мышиные линии К+АТФ-нокаут и К+АТФ-LOF (утрата функции, loss-of-function)) отмечалось сохранение уровня секреции инсулина [5]. Терапия глибенкламидом, приводящая к увеличению возбудимости клеточных мембран, не вела к какому-либо значительному снижению уровня инсулина в островках поджелудочной железы на моделях in vivo [6].
Такие данные подтверждают преимущественную роль дедифференцировки β-клеток поджелудочной железы в сторону клеток-предшественниц при тяжелом течении сахарного диабета. Вектор дифференцировки изменяется обратно в сторону зрелых β-клеток после снижения уровня глюкозы в крови путем интенсивной инсулинотерапии [7].
Различия in vivo и in vitro могут возникать из-за того, что продолжительность воздействия высокого уровня глюкозы in vitro недостаточна для возникновения долгосрочных последствий, обнаруживаемых in vivо. Кроме того, отсутствие in vitro других активных веществ, таких как аминокислоты и гормоны инкретины (модуляторы чувствительности β-клеток к уровню глюкозы), также изменяет клеточный ответ [5].
Стрессовые эффекты, которым β-клетки подвергаются in vitro (например, в ходе выделения островков: нарушение их васкуляризации, потеря эндотелиальных клеток и изменения внутриклеточного матрикса), отсутствуют in vivo [5].
Интересен вопрос влияния экзогенного инсулина на функции β-клеток. Логично, что добавление инсулина в инкубационную среду способно защитить клетки островков от потери секреции эндогенного инсулина. Эти данные согласуются с исследованиями, в которых продемонстрировано сохранение функции β-клеток у пациентов с недавно диагностированным сахарным диабетом 2 типа, подвергшихся интенсивной терапии инсулином, по сравнению с пациентами, получавшими сульфонилмочевину [8, 9].
Предполагается, что обработка экзогенным инсулином (а также инсулином с диазоксидом) приводит к условному «покою» β-клеток, что позволяет восстановить клеточную функцию. Клетка получает отдых от гиперстимулированного метаболизма, гипервозбуждения, повышенного уровня ионов кальция и, конечно, от необходимости самостоятельной секреции инсулина. Но, хотя отдых от секреции является самым очевидным и закономерным, он не является определяющим фактором. Экспериментально выявлено, что именно отдых от повышенной возбудимости (и его последствий) является ключевым аспектом, влияющим на содержание инсулина, в изолированных условиях (in vitro) [5, 10].
Такие результаты можно экстраполировать на общую концепцию развития сахарного диабета. Возможно, именно гиперполяризация мембран β-клеток на ранних стадиях заболевания определяет дальнейшее его прогрессирование ввиду потери клеточной функции. И терапевтическая коррекция ионных каналов способна при ранней диагностике значительно изменить прогноз для пациента и сохранить активную клеточную массу островков Лангерганса.
.
Источники:
1. Emfinger C. H. et al. Expression and function of ATP-dependent potassium channels in zebrafish islet beta-cells. R Soc Open Sci.2017;4:160808.
2. Remedi M. S., Emfinger C. Pancreatic beta-cell identity in diabetes. Diabetes Obes Metab.2016;18(Suppl 1):110–116.
3. Glynn E. et al. Chronic Glucose Exposure Systematically Shifts the Oscillatory Threshold of Mouse Islets: Experimental Evidence for an Early Intrinsic Mechanism of Compensation for Hyperglycemia. Endocrinology.2016;157:611–623.
4. Cinti F. et al. Evidence of beta-cell Dedifferentiation in Human Type 2 Diabetes. The Journal of clinical endocrinology and metabolism. 2015;101:1044–1054.
5. Shyr Z.A., Wang Z., York N.W., Nichols C.G., Remedi M.S. The role of membrane
excitability in pancreatic β-cell glucotoxicity. Sci Rep. 2019;9(1):6952.
6. Remedi M. S., Nichols C. G. Chronic antidiabetic sulfonylureas in vivo: reversible effects on mouse pancreatic beta-cells. PLoS Med.2008;5:e206.
7.Wang, Z., York, N. W., Nichols, C. G. & Remedi, M. S. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab.2014;19:872–882.
8. Wajchenber B. L. beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev.2007;28:187–218.
9. Alvarsson M. et al. Effects of insulin vs. glibenclamide in recently diagnosed patients with type 2 diabetes: a 4-year follow-up. Diabetes Obes Metab.2008;10:421–429.
10. Alarcon C. et al. Pancreatic beta-Cell Adaptive Plasticity in Obesity Increases Insulin Production but Adversely Affects Secretory Function. Diabetes.2016;65:438–450.
