
Витаминотерапия как этиотропная помощь при наследственных неврологических заболеваниях взрослого возраста
Неврологические генетические заболевания, манифестирующие в подростковом и взрослом возрасте, принадлежат к гетерогенным состояниям, обусловленным нарушением множества генов и проявляющимися крайне разнообразными фенотипами. Зачастую такие заболевания не имеют специфического лечения, однако некоторые из этих заболеваний поддаются терапии витаминами [1]. Витамины — необходимые микронутриенты, синтезируемые в организме в недостаточных количествах либо вовсе не синтезируемые, тем не менее, играющие важнейшую роль во внутриклеточной машинерии, трансформируясь в активные формы — кофакторы ферментов.
Витаминные добавки легкодоступны, обычно не вызывают выраженных побочных эффектов и эффективны, особенно на ранних этапах заболеваний. Таким образом, своевременное распознавание витамин-связанных заболеваний критически важно для неврологов.
Эта статья направлена на описание фенотипов витамин-зависимых нейрогенетических заболеваний с манифестацией в подростковом и взрослом возрасте, явно отличающихся от раннего дебюта.
Методы
Авторами статьи было определено 24 нейрогенетических заболевания, неврологические проявления которых могут возникать в возрасте от 10 лет, и характеризуются видимой реакцией на прием витаминных добавок, по крайней мере, для части пациентов. Описываются следующие витамины: A, B1, B2, B3, B6, B8, B9, B12, E и тетрагидробиоптерин (BH4) коэнзим. Авторы выбрали именно этот возрастной порог (упоминаемый в тексте как «взрослый возраст»), поскольку, по опыту авторов, наследственные метаболические заболевания, начинающиеся в возрасте старше 10 лет, имеют фенотипы, которые похожи на фенотипы пациентов с более поздним дебютом этих заболеваний и отличаются от форм данной патологии с более ранним началом [2–4]. Несмотря на то, что BH4 не является полноценным витамином, он представляет собой органическое соединение, действующее аналогично витаминным кофакторам [5], что объясняет решение авторов добавить BH4-ассоциированные состояния к заболеваниям, чувствительным к витаминам [6].
Обзор
Нейрогенетические заболевания, отвечающие на прием витаминов были поделены авторами статьи на 4 группы:
- нарушение синтеза кофакторов, образующихся из витаминов;
- нарушение транспорта (абсорбция в ЖКТ, транспорт в крови или захват клетками);
- дефект самого энзима, кофактор которого синтезируется из витаминов;
- вторичный витаминный дефицит (аномалии, не ассоциированные с генетическими расстройствами, вовлеченными в метаболизм витаминов).
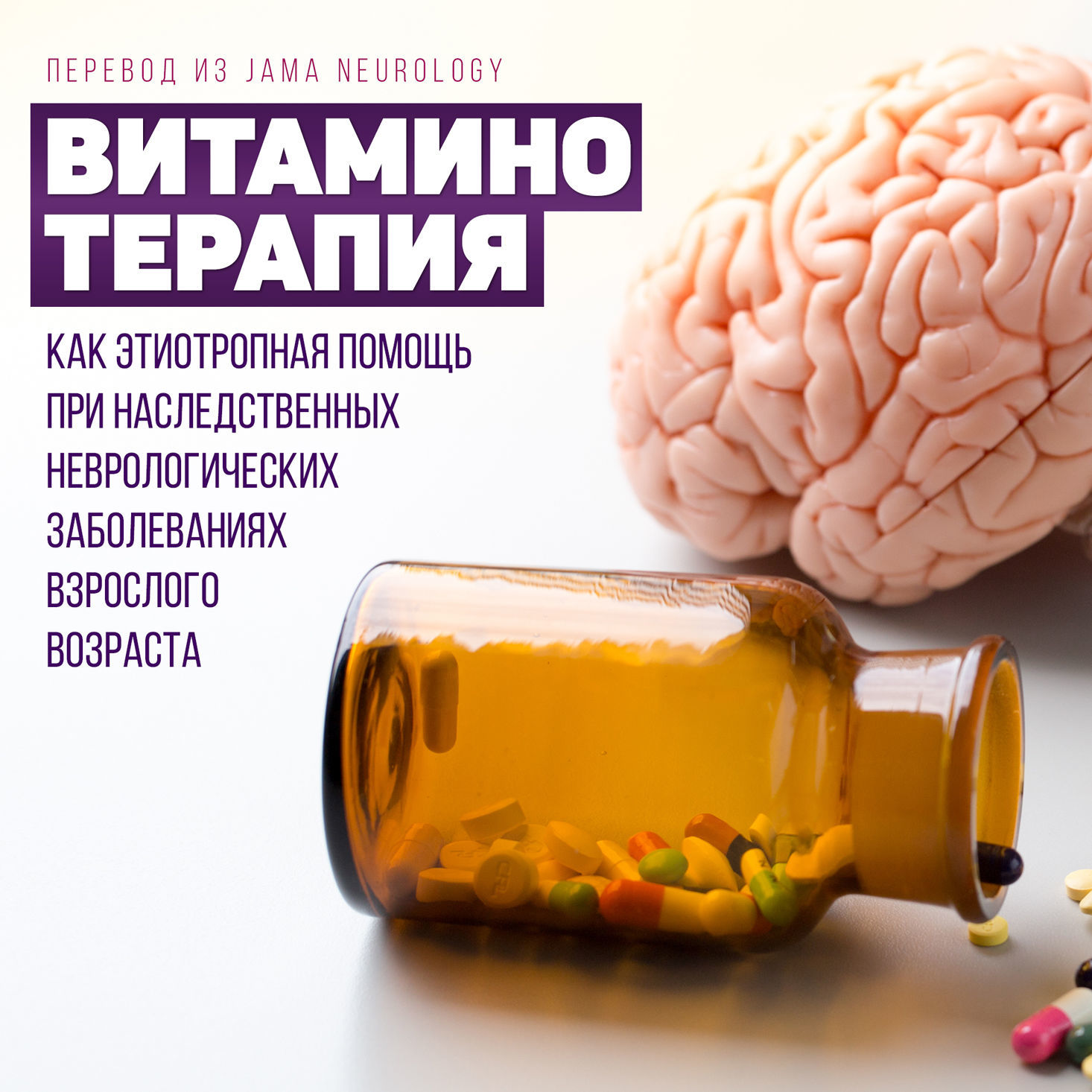
Рисунок 1 демонстрирует групповую принадлежность на примере метаболизма кобаламина.
Двойные стрелки указывают на нарушение активности PLP при накоплении P5C и P6C.
В зависимости от патофизиологии, для оптимальной эффективности применения витаминов они (или синтезируемые из них кофакторы) могут назначаться в различных фармакологических дозах и с различными путями введения. В случае протекания заболевания по вышеуказанному третьему типу, эффективность зависит от этапа вовлечения последовательных витамин-связанных реакций, часто выявляемых среди форм болезни, манифестирующих во взрослом возрасте [7–10], тогда как в случае 4 типа эффективность скорее будет слабо выражена, поскольку недостаток витаминов может представлять собой лишь эпифеномен заболевания.
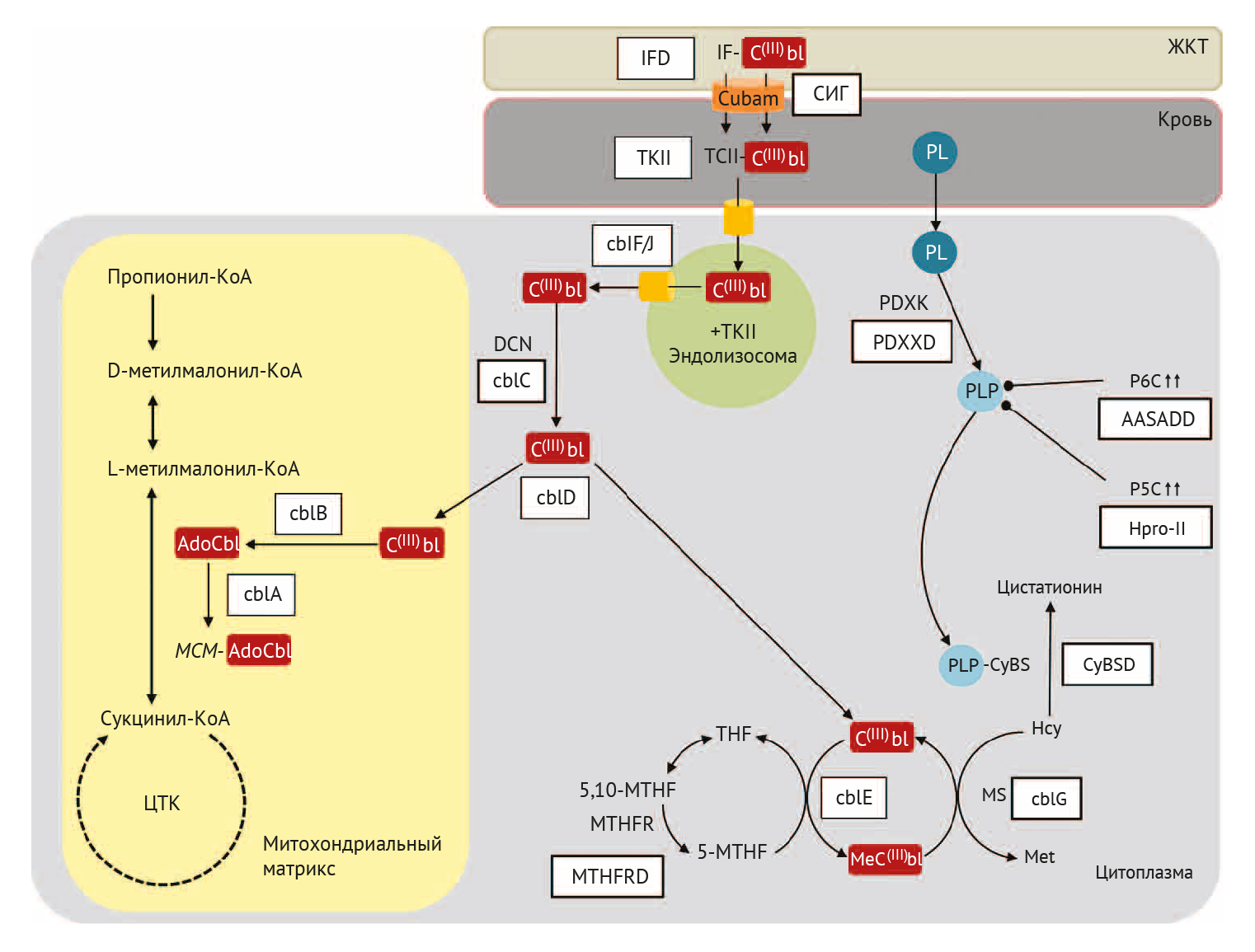
Основываясь на том, что большинство фенотипов проявляются в результате различных генетических заболеваний, для более прагматичного взгляда к этим многочисленным и сложным состояниям выделяют клинико-биологический подход. Собранные данные по некоторым нейрогенетическим заболеваниям представлены на рисунке 2.
5-MTHFR, 5-метилтетрагидрофолат; AASA, α-аминоадипин-δ-полуальдегид; ABL, абеталипопротеинемия; ACAD9, ацил-коА-дегидрогеназа 9; ACAD9D, дефицит ацил-коА дегидрогеназы 9; AIFM1, ген фактора индукции апоптоза, ассоциированный с митохондриями 1; AMN, трансмембранный белок, связанный с амнионом; ТТПА, ген белка-переносчика α-токоферола; AVED, атаксия с дефицитом витамина E; BCAT2, ген трансаминазы 2 аминокислот с разветвленной цепью; BCAT2D, дефицит трансаминазы 2 аминокислот с разветвленной цепью; BiD, дефицит биотинидазы; BTD, ген биотинидазы; BTRE, биотин-тиамин-зависимая энцефалопатия; CBS, ген цистатионин-β-синтазы; CFD, церебральный дефицит фолиевой кислоты; CMT, Шарко — Мари — Тута; CMTX4, Х-сцепленная болезнь Шарко — Мари — Тута 4 типа; CSF, спинномозговая жидкость; CUBN, ген кубилина; CyBSD, дефицит цистатионин-β-синтазы; DNAJC12, ген C12, член семейства белков теплового шока DnaJ; ETFA/ETFB/ETFDH, гены флавопротеинов, переносящих электроны, альфа, бета и дегидрогеназы; FLAD1, ген флавинадениндинуклеотидсинтетазы 1; GI, желудочно-кишечный тракт; HD, болезнь Хартнупа; HPNBH4, гиперфенилаланинемия, легкая, без дефицита BH4; IGS, синдром Имерслунда — Грасбека; LSMFLAD, миопатия накопления липидов вследствие дефицита синтетазы FAD; MADD, множественный дефицит ацил-коА; MMACHC, метаболизм гена C, связанного с кобаламином; МRI, магнитно-резонансная томография; MTR, ген 5-метилтетрагидрофолат-гомоцистеинметилтрансферазы; MTHFR, метилентетрагидрофолатредуктаза; MTRFRD, дефицит метилентетрагидрофолатредуктазы; MTTP, ген белка-переносчика микросомальных триглицеридов; P6C, Δ1-пиперидин-6-карбоксилат; PDE, пиридоксинзависимые эпилепсии; PDHA1, ген субъединицы E1-α пируватдегидрогеназы; PDHAD, дефицит E1-α пируватдегидрогеназы; PDXK, пиридоксалкиназа; PDXKD, дефицит PDXK; PAH, ген фенилаланингидроксилазы; PKU, фенилкетонурия; PLP, пиридоксаль-5'-фосфат; RREI, непереносимость физических упражнений, реагирующих на рибофлавин; RTD, дефицит транспортера рибофлавина; SLC19A3, семейство 19 носителей растворенного вещества, член 3; SLC6A19, семейство 6 носителей растворенного вещества, член 19; SLC52A2/SLC52A3, семейство 52 носителей растворенного вещества, члены 2 и 3; SLC25A32, семейство 25 носителей растворенного вещества, член 32.
Расстройства метаболизма тиамина и поражение базальных ганглиев
Энцефалопатия Вернике — это приобретенное заболевание, вызванное дефицитом тиамина (витамина B1), часто в контексте недоедания и/или злоупотребления алкоголем. Энцефалопатия обычно сопровождается мозжечковыми нарушениями, судорогами, офтальмоплегией и/или птозом [11]. Схожий фенотип можно встретить при двух тиамин-ассоциированных проявляющихся во взрослом возрасте нейрогенетических заболеваниях: биотин-тиамин-зависимой энцефалопатии (BTRE) и дефиците E1-α пируватдегидрогеназы (PDHAD). Эти два состояния являются поддающимися лечению причинами генетически гетерогенного синдрома Лея (Ли) [12]. При BTRE транспорт тиамина в цитозоль снижается в результате дефекта переносчика тиамина 2-го типа (кодируется геном семейства растворенных носителей-19, член 3 [SLC19A3]), что приводит к низкому внутриклеточному уровню тиамина, тогда как уровень сывороточного тиамина остается в норме [9]. В случае PDHAD аномальная субъединица E1-α пируватдегидрогеназы (кодируемая геном субъединицы E1-α пируватдегидрогеназы [PDHA1]) препятствует правильному связыванию его кофактора тиаминпирофосфата, активного тиамина (рис. 3) [13]. Этот сбой оказывает влияние на цикл трикарбоновых кислот с уменьшением выработки ацетил-КоА и приводит к переключению на анаэробное производство энергии и, в конечном итоге, к лактоацидозу, который также присутствует при BTRE. Помимо высокого уровня лактата, характерными чертами PDHAD являются высокие уровни пирувата в крови и спинномозговой жидкости. Как и при других метаболических заболеваниях, стрессовые состояния, такие как приступы лихорадки, хирургическое вмешательство или травмы, могут вызвать эпизоды энцефалопатии. Однако также могут возникать прогрессивно ухудшающиеся хронические симптомы, такие как дистония (также присутствующая в острой фазе). МРТ головного мозга обычно показывает Т2-гиперинтенсивный сигнал от базальных ганглиев (хвостатых ядер и скорлупы) (рис. 4 [A и B при BTRE]) в случае синдрома Лея и от срединных структур таламуса и околоводопроводного серого вещества в случае энцефалопатии Вернике [14]. При BTRE в острых фазах заболевания было описано поражение коры головного мозга с рассеянными гиперинтенсивными по Т2 очагами [15,16]. Несмотря на то, что тиамин является основой лечения обоих заболеваний, при BTRE также рекомендуется принимать добавки с биотином, которые, вероятно, повышают экспрессию SLCA19A3 посредством биотинилирования гистонов [17]. Кетогенная диета назначается при PDHAD, поскольку она увеличивает выработку печенью кетоновых тел, альтернативного глюкозе энергетического субстрата для образования ацетил-КоА в центральной нервной системе [18].
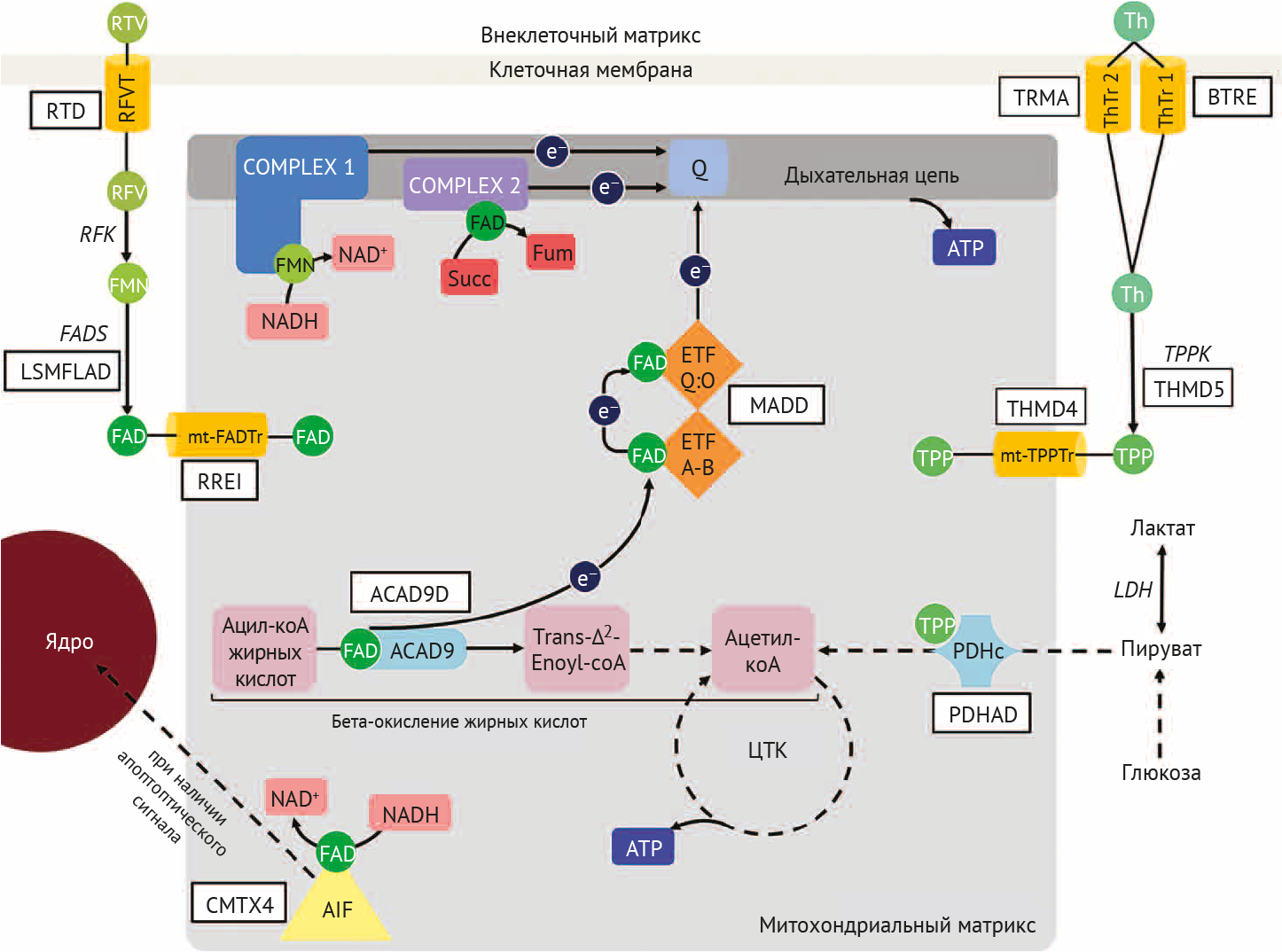
Q, убихинон; RFV, рибофлавин; RFVT, переносчик рибофлавина; RFK, рибофлавинкиназа; Succ, сукцинат; THMD4, чувствительная к тиамину двусторонняя дегенерация полосатого тела и полинейропатия; THMD5, энцефалопатия из-за недостаточности тиаминпирофосфокиназы; ThTr-1, переносчик тиамина-1; TPP, пирофосфат тиамина, активная форма тиамина; ТППК, тиаминпирофосфокиназа; TRMA, тиамин-чувствительная мегалобластная анемия.
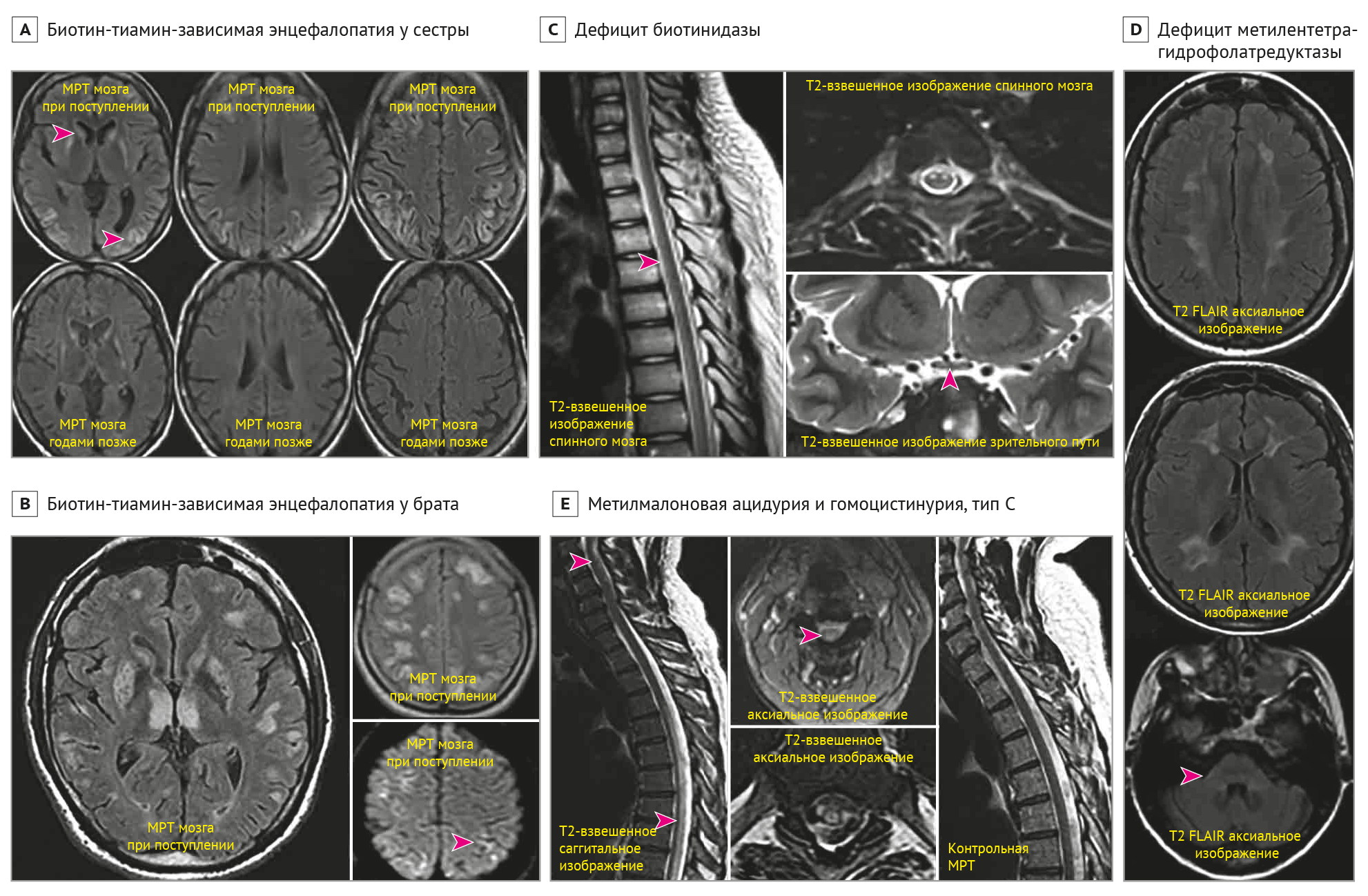
A | МРТ головного мозга сестры демонстрирует двустороннюю гиперинтенсивность головок хвостатых ядер (верхняя стрелка) и скорлупы, а также кортикальные и юкстакортикальные поражения белого вещества (нижняя стрелка) с двухсторонним гиперинтенсивным сигналом по Т2-FLAIR. Несколько лет спустя контрольная МРТ (изображения в нижнем ряду) показала почти полный регресс поражений. Вместо этого стала заметна серьезная диффузная атрофия мозга.
B | У брата была энцефалопатия по типу Вернике с симметричными измененными по T2-FLAIR сигналами от таламуса, наряду с гиперинтенсивностью базальных ганглиев и диффузными кортикальными и юкстакортикальными поражениями белого вещества. DWI-изображения демонстрируют некоторые тонкие очаги гиперинтенсивности, в основном внутри кортикальных поражений (стрелка).
C | Дефицит биотинидазы; Т2-ВИ МРТ-изображения спинного мозга и зрительного пути. Визуализируются протяженные и обширные Т2-гиперинтенсивные очаги спереди и билатерально в спинном мозге (стрелка). Гиперинтенсивный Т2-сигнал от хиазмы (стрелка).
D | Дефицит метилентетрагидрофолатредуктазы; аксиальная последовательность T2-FLAIR показывает симметричную и двустороннюю обширную лейкоэнцефалопатию с сохранением U-волокон. Подобное изменение сигнала было заметно в средних ножках мозжечка.
E | Метилмалоновая ацидурия и гомоцистинурия, тип cblC; Т2-взвешенные сагиттальные и аксиальные срезы показывают обширное поражение в основном задней части спинного мозга (стрелки). Контрольная МРТ показала почти полный регресс поражений на Т2-ВИ.
Рибофлавин в мышцах и заболевания нервной системы
Рибофлавин (или витамин B2) — водорастворимое и светочувствительное соединение, являющееся источником флавинмононуклеотида и флавинадениндинуклеотида (FAD). Эти два кофактора действуют как переносчики электронов во многих биохимических реакциях, в том числе в дыхательной цепи митохондрий, с участием белков, называемых электрон-переносящими флавопротеинами (ETF). Приобретенная недостаточность рибофлавина встречается редко и может быть связана с неправильным питанием (особенно при низком потреблении молока), злоупотреблением алкоголем (этанол блокирует рецепторы рибофлавина, нарушая его всасывание из кишечника) и фототерапией у детей (используется для лечения гипербилирубинемии) [19]. Описанные здесь генетические заболевания, в развитии которых играет роль рибофлавин, относятся к нарушениям (1) внутриклеточной его доставки при дефекте транспортера рибофлавина (RTD), (2) митохондриального β-окисления жирных кислот, наблюдаемого при множественном дефиците ацил-КоА (MADD; дефекты ETF и ETF убихинон-оксидоредуктазы), довольно похожие недавно описанные заболевания (дефицит ацил-коА-дегидрогеназы 9, болезнь накопления липидов с миопатией, связанная с дефицитом синтетазы FAD, и непереносимость упражнений в ответ на прием рибофлавина, приписываемая дефекту митохондриального транспортера FAD); а также (3) FAD-зависимая оксидоредуктаза при Х-сцепленной болезни Шарко — Мари — Тута 4 типа (CMTX4) (рис. 3). При всех этих состояниях характерной чертой для манифестации во взрослом возрасте является патология периферических нервов или мышц. Таким образом, пациенты с RTD имеют чисто моторную (реже сенсомоторную) нейронопатию с ранними бульбарными симптомами, что может напоминать боковой амиотрофический склероз; пациенты с CMTX4 демонстрируют синдром Шарко — Мари — Тута-подобное заболевание (наследственная дистальная сенсомоторная периферическая нейропатия) с двигательными нарушениями, дистальной амиотрофией и слабостью, арефлексией и «полыми» стопами (pes cavus). При позднем дебюте MADD обычно проявляется миопатией в виде слабости проксимальной мускулатуры и непереносимости физических упражнений. Интересно, что как при RTD, так и при CMTX4 на ранней стадии заболевания выявляется нейросенсорная тугоухость. Также оптическая нейропатия может быть связана с наличием у пациентов RTD. Множество заболеваний, патогенез которых приводит к дефициту ацил-коА, могут также проявляться острой декомпенсацией с лактоацидозом и рабдомиолизом в случаях повышенной потребности в энергии (например, интенсивные физические упражнения, голодание, инфекции), чрезмерного употребления алкоголя, рвоты или диареи. Метаболические исследования могут помочь в диагностике MADD и связанных с ним заболеваниях, при которых профиль ацилкарнитина в крови всегда нарушен и характеризуется накоплением полноцепочечных ацилкарнитинов, тогда как эта особенность не является постоянной при RTD. Добавки рибофлавина обычно приводят к быстрому и значительному улучшению при MADD и связанных с ним заболеваниях, тогда как клинические эффекты при RTD и CMTX4 обычно проявляются в течение нескольких месяцев.
Разнообразные и комплексные клинические проявления дефектов обмена пиридоксина, фолата и кобаламина
Пиридоксин, фолат и кобаламин, также называемые B6, B9 и B12 соответственно, рутинно измеряются в крови. Их дефицит может восполняться, о чем говорит частый регресс неврологической симптоматики на фоне их приобретенного дефицита [20]. Пиридоксин действует как кофактор фермента цистатионин-β-синтазы (CyBS), который обеспечивает трансульфурацию гомоцистеина (Hcy) в цистатионин для его разложения. Фолат и Cbl также непосредственно участвуют в метаболизме Hcy, причем они оба необходимы для его реметилирования до Met (рис. 1). Следовательно, генетический дефект биохимического пути каждого из этих витаминов может привести к тяжелой гипергомоцистеинемии (обычно более 100 мкмоль/л), связанной с высокими уровнями Met в крови у лиц с дефицитом CyBS или низкими уровнями Met у лиц с фолат и/или Cbl-ассоциированными заболеваниями. С другой стороны, концентрация витаминов в крови часто остается в норме. Пиридоксин также участвует во многих других ферментативных реакциях (включая выработку нейротрансмиттеров, таких как γ-аминомасляная кислота), что объясняет очень разнообразные проявления нейрогенетических заболеваний, связанных с пиридоксином среди взрослых: цереброваскулярный тромбоз в молодом возрасте (< 50 лет) при дефиците CyBS, ассоциированный с эктопией хрусталика глаза и марфаноидным габитусом, эпилепсия с множественной лекарственной резистентностью при пиридоксин-зависимой эпилепсии, тяжелая лейкоэнцефалопатия с неспецифическими легкими симптомами при дефиците трансферазы аминокислот с разветвленной цепью 2 и Шарко — Мари — Тута-подобная аксональная периферическая нейропатия с двусторонней оптической нейропатией при недостаточности пиридоксалькиназы. Несмотря на то, что пиридоксин остается основой лечения этих состояний, дозы более 200 мг/день могут вызывать серьезные побочные эффекты, наиболее важным из которых является сенсорная нейронопатия [21].
Чрезмерное потребление фолиевой кислоты (например, во время беременности) или ее приобретенный дефицит, обычно связанный с неправильным питанием и/или приемом лекарств (например, метотрексата), могут привести к сложной клинической картине, что затрагивает как центральную, так и периферическую нервную систему с острыми и/или хроническими когнитивными, психиатрическими и/или двигательными проявлениями, связанными с лейкоэнцефалопатией (рис. 4D), миелопатией (иногда с гиперинтенсивностью на Т2 МРТ) и/или аксональной полинейропатией [22]. Наиболее частый генетический дефект метаболизма фолиевой кислоты — недостаточность 5,10-метилентетрагидрофолатредуктазы — может представлять этот широкий и гетерогенный фенотип. Недавно был описан другой синдром у взрослых, также связанный с метаболизмом фолиевой кислоты, называемый церебральной фолиевой недостаточностью. Он проявляется низким уровнем фолиевой кислоты в спинномозговой жидкости в результате нарушения транспорта активной формы фолиевой кислоты из крови в спинномозговую жидкость через ГЭБ. Дефицит церебрального фолата встречается в основном при митохондриальных нарушениях, но основная часть церебрального дефицита фолиевой кислоты, возникающая на позднем этапе, несмотря на подозрения о генетическом происхождении заболевания, в настоящее время остается невыясненной [23]. Помимо метаболизма Hcy, Cbl также участвует в производстве сукцинил-КоА, промежуточного продукта цикла Кребса, производного от метилмалонил-КоА (рис. 1). Метилмалоновая ацидурия и гомоцистинурия типа C — наиболее распространенные генетические дефекты метаболизма Cbl, описанные у взрослых, характеризующиеся повышенными уровнями метилмалоновой кислоты и Hcy. Поздний дебют cblC характеризуется схожим с дефицитом 5,10-метилентетрагидрофолатредуктазы клинико-радиологическим фенотипом (рис. 4E), за исключением возможного развития гемолитико-уремического синдрома и/или легочной артериальной гипертензии. Поскольку cblC обусловлен дефектом децианирования Cbl, цианокобаламин (классическая лекарственная форма Cbl) использовать нельзя, и пациентов следует лечить гидроксикобаламином. У взрослых были описаны два других, менее частых дефекта метаболизма Cbl: cblG, вызванный дефицитом метионинсинтазы, ответственной за реметилирование Hcy до Met, и синдром Имерслунда — Гресбека, при котором нарушение транспортировки из ЖКТ приводит к дефициту Cbl в крови.
Болезнь Хартнупа, генетический симулякр пеллагры
Ниацин, или витамин B3, дефицит которого приводит к развитию пеллагры, в настоящее время в развитых странах считается редким заболеванием [24]. Болезнь Хартнупа — генетически детерминированное расстройство, вызванное мутациями гена, кодирующего транспортер нейтральных аминокислот B0AT1, в основном экспрессируемого в кишечнике и проксимальных канальцах почек, имеет почти идентичную пеллагре клиническую картину, проявляющуюся мозжечковой атаксией, психиатрической симптоматикой и фотофобией. Фактически нарушение этого переносчика приводит к потере нескольких нейтральных аминокислот, включая триптофан, который является не только предшественником ниацина, но также участвует в производстве мелатонина и серотонина.
Дефицит биотинидазы: имитируя нейрооптикомиелит
Биотинидаза — это фермент, отвечающий за переработку биотина (витамина B8), кофактора карбоксилаз человека. Дефицит биотинидазы, таким образом, приводит к дефициту биотина, снижению активности карбоксилаз и, как следствие, нарушению некоторых метаболических функций, таких как глюконеогенез, биосинтез жирных кислот и катаболизм аминокислот. Как и при других энергетических дефектах (например, BTRE, PDHAD, MADD), симптомы часто усугубляются стрессовым событием (например, инфекцией, хирургической операцией или травмой). С клинической точки зрения, поздние формы проявляются двусторонним оптическим невритом и миелитом, вовлекающим три и более сегментов спинного мозга [25] (рис. 4C), что включает это заболевание в дифференциальную диагностику в случаях серонегативного или рефрактерного к терапии оптиконевромиелита.
Витамин Е и эквилибриум
Все витамины, описанные выше, являются водорастворимыми, но четыре — жирорастворимые: витамин А (ретинол), три формы витамина К (филлохинон, менахинон и менадион), витамин D (холекальциферол) и витамин E (токоферол). Среди них неврологические расстройства у взрослых формируются в результате дефицита витаминов А или Е. Недостаток этих двух витаминов в настоящее время встречается редко, но может быть вызван нарушением липидного обмена и/или всасывания.
Например, абеталипопротеинемия (ABL) представляет собой генетический дефект микросомального белка-переносчика триглицеридов, основная функция которого заключается в переносе последних на аполипопротеин B для образования липопротеинов очень низкой плотности (ЛПОНП) в гепатоцитах и хиломикронов в энтероцитах [26]. Таким образом, нарушается синтез хиломикронов и ЛПОНП, переносящих жирорастворимые витамины в крови, что приводит к снижению транспорта этих витаминов в периферические ткани. Другое заболевание, в которое вовлечено нарушение обмена только витамина E, называется атаксией с дефицитом витамина E; оно вызвано дефектом белка-переносчика αльфа-токоферола (α-TTP), который отвечает за включение витамина в ЛПОНП, приводя к его деградации [27]. Как при ABL, так и при атаксии с дефицитом витамина E, уровни витамина E в крови очень низкие, и пациенты страдают от двигательных нарушений в виду мозжечковой и сенситивной атаксии (из-за поражения задних столбов и/или сенсорной нейронопатии). Кроме того, у пациентов с ABL синдром мальабсорбции (т. е., хроническая диарея, низкий рост, анемия), начинающийся в детстве [28, 29], и никталопия (ночная слепота, прим. переводчика), которая является известным следствием недостаточности витамина A для функции сетчатки, [30] часто проявляются в дебюте заболевания. Когда лечение начинается на этом этапе, оно обычно помогает предотвратить неврологические нарушения [31]. При атаксии с дефицитом витамина E его добавление может улучшить симптомы, если его принимать на ранней стадии заболевания [27]. Важно отметить, что хотя витамин A следует вводить при ABL, дозы более 4000 МЕ/кг для взрослых могут вызывать побочные эффекты, такие как поражения кожи, анемия и гиперкальциемия, тогда как дозы более 10 000 МЕ/день у беременных являются тератогенными для плода [32].
BH4 и паркинсонизм
Паркинсонизм — это клиническое проявление низкого уровня в мозге нейромедиатора дофамина (грубое обобщение, прим. переводчика), синтезируемого из фенилаланина в ходе трехэтапного синтетического пути с участием ферментов фенилаланингидроксилазы и тирозингидроксилазы, для которых требуется BH4 в качестве кофактора и молекула шаперона, белок теплового шока DNAJC12 [33]. Фенилкетонурия, вызванная нарушением функции фенилаланингидроксилазы, является распространенной генетической причиной когнитивных нарушений, которые можно профилактировать с помощью низкобелковой диеты, часто применяемой после скрининга новорожденных. Фенилкетонурия иногда показывает клинико-биохимический ответ на введение BH4. Редко, начиная с подросткового или взрослого возраста, клинические проявления включают паркинсонизм, часто ассоциированный с потерей остроты зрения, когнитивными нарушениями и/или лейкоэнцефалопатией. Пациенты, отвечающие и не отвечающие на терапию, имеют схожий фенотип, но разные генотипы [34]. Другое недавно обнаруженное состояние, вызванное вариациями последовательности в гене, кодирующего один из членов семейства белков теплового шока DnaJ C12 (DNAJC12), вызывает непрогрессирующий паркинсонизм и легкие когнитивные нарушения у взрослых с хорошей чувствительностью к терапии BH4 [35].
Ограничения
Кофакторы неорганических ферментов (в основном микроэлементы) выходят за рамки настоящего обзора. Ассоциированные заболевания были описаны в других источниках [36–40].
Заключение
Нейрогенетические витамин-зависимые заболевания встречаются редко, что, вероятно, связано с гиподиагностикой. Более глубокие клинические знания этого раздела могут помочь неврологу определить диагноз и назначить лечение, значительно улучшающее качество жизни пациентов. Клинически ориентированные генетические панели, полноэкзомное и полногеномное секвенирование теперь являются доступными инструментами для облегчения процесса диагностики. Однако в некоторых клинических случаях, описанных в этом обзоре, клинико-биохимический подход все еще применим по нескольким причинам. Во-первых, определенные клинические случаи изначально не предполагают генетическую причину, что побуждает к исчерпывающим исследованиям и поиску механизма нарушенного обмена. Во-вторых, результаты генетического тестирования могут отсрочить введение витаминов в ситуациях, когда важно назначить раннее лечение. В-третьих, генетическое тестирование может пропустить некоторые варианты, которые трудно идентифицировать или интерпретировать, особенно при секвенировании всего экзома и генома. Наконец, генетические тесты все же дороже и не так широко распространены, как витаминные добавки. Поскольку эта область быстро расширяется, вполне вероятно, что в будущем среди взрослых будут выявляться некоторые заболевания, чувствительные к приему витаминов, иногда с неожиданным фенотипом, отличным от манифестации в детском возрасте. Также часто регистрируются новые нейрометаболические заболевания, связанные с нарушением обмена витаминов. По этой причине авторы статьи предлагают рассмотреть возможность исследования приема витаминных добавок у пациентов с фенотипами, сходными с известными генетическими заболеваниями, чувствительными к витаминам, даже если биохимические и генетические исследования (в том числе полногеномное секвенирование) дали отрицательные результаты.
