
Лизосомные болезни накопления

Данная статья написана с целью ознакомить читателя с лизосомными болезнями накопления и дать их краткое описание, разобрав пять наиболее распространенных из них. Невозможно в один обзор объединить полное описание каждого заболевания, но мы надеемся, что прочтение данного материала подтолкнет Вас к дальнейшему изучению этой группы патологии.
На данный момент насчитывается около 70 лизосомных болезней накопления (ЛБН). В основном они обусловлены дефектом фермента, в меньшей степени — транспортера или ассоциированного с ним белка. ЛБН развиваются в результате точечных мутаций: миссенс и нонсенс, зачастую имеющих аутосомно-рецессивный либо X-сцепленный (в случае болезней Данона, Фабри, Хантера) тип наследования, а также изредка могут возникать спорадически (de novo). Миссенс мутации характеризуются тем, что кодон начинает кодировать другую аминокислоту, но в некоторых случаях это не влияет на функцию белка или влияет незначительно. Нонсенс мутации же приводят к появлению стоп-кодона, который прерывает дальнейший синтез белка [1].
Суммарно ЛБН встречаются в 1 случае на 5 тыс. человек, однако каждая патология в отдельности — крайне редкое явление. Тем не менее это зависит от точки зрения. Для генетически детерминированных ЛБН характерно свойство выявляться с большей частотой в определенных группах. Наиболее распространенные из них — болезнь Фабри (до 2,5 на 100 тыс. мужчин), болезнь Гоше (до 2 на 100 тыс. человек), метахроматическая лейкодистрофия (до 2,5 на 100 тыс. человек) и болезнь Помпе (до 2,5 на 100 тыс. человек). В таблице 1 приведена частота выявления отдельных ЛБН среди определенных этносов, что, вероятно, связано с эффектом основателя (явление снижения и смещения генетического разнообразия при заселении малым количеством представителей рассматриваемого вида новой географической территории [2]).
Таблица 1. Распространенность ЛБН в зависимости от этноса
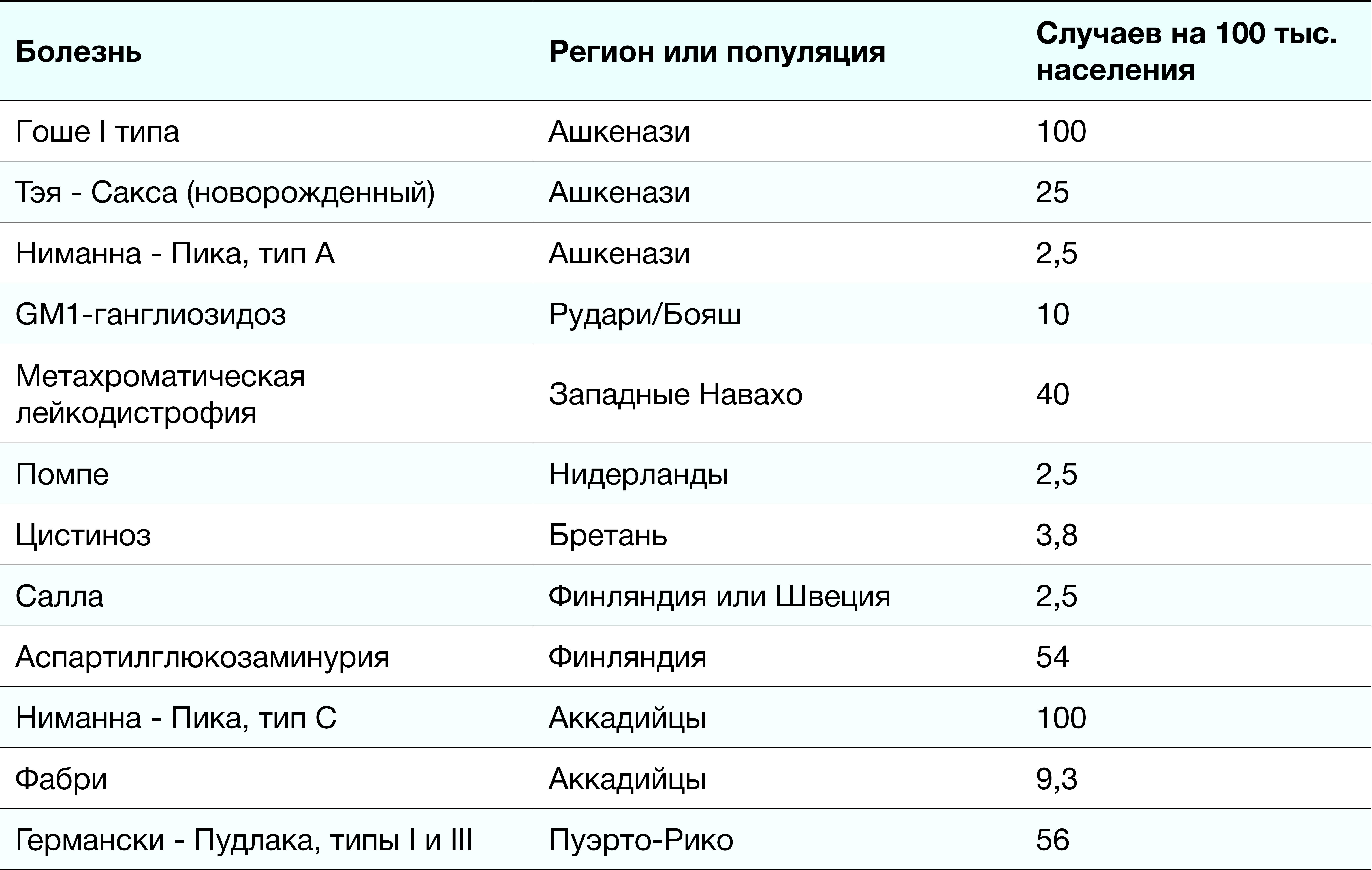
Таблица 2. Краткое описание ЛБН [1]
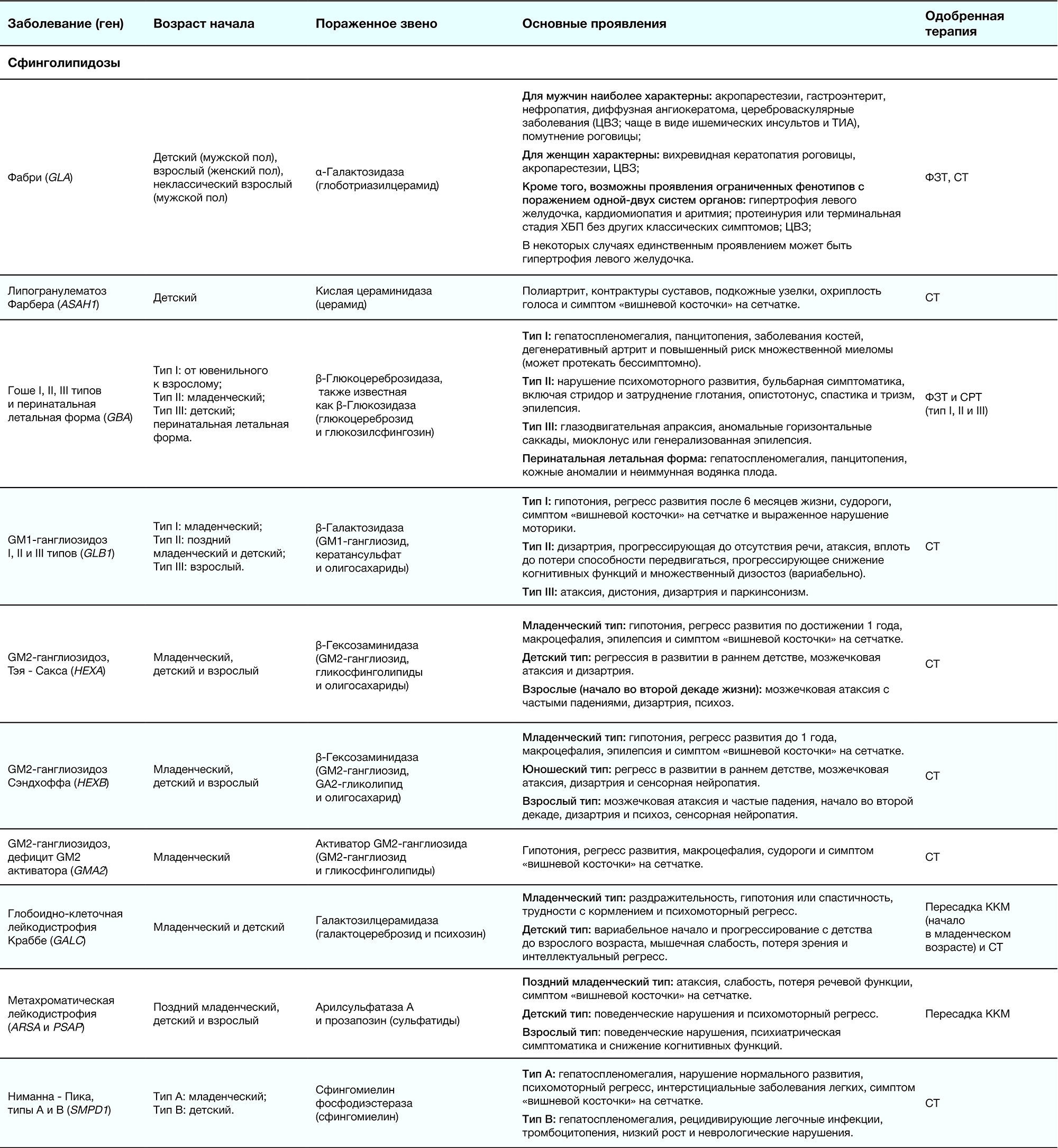
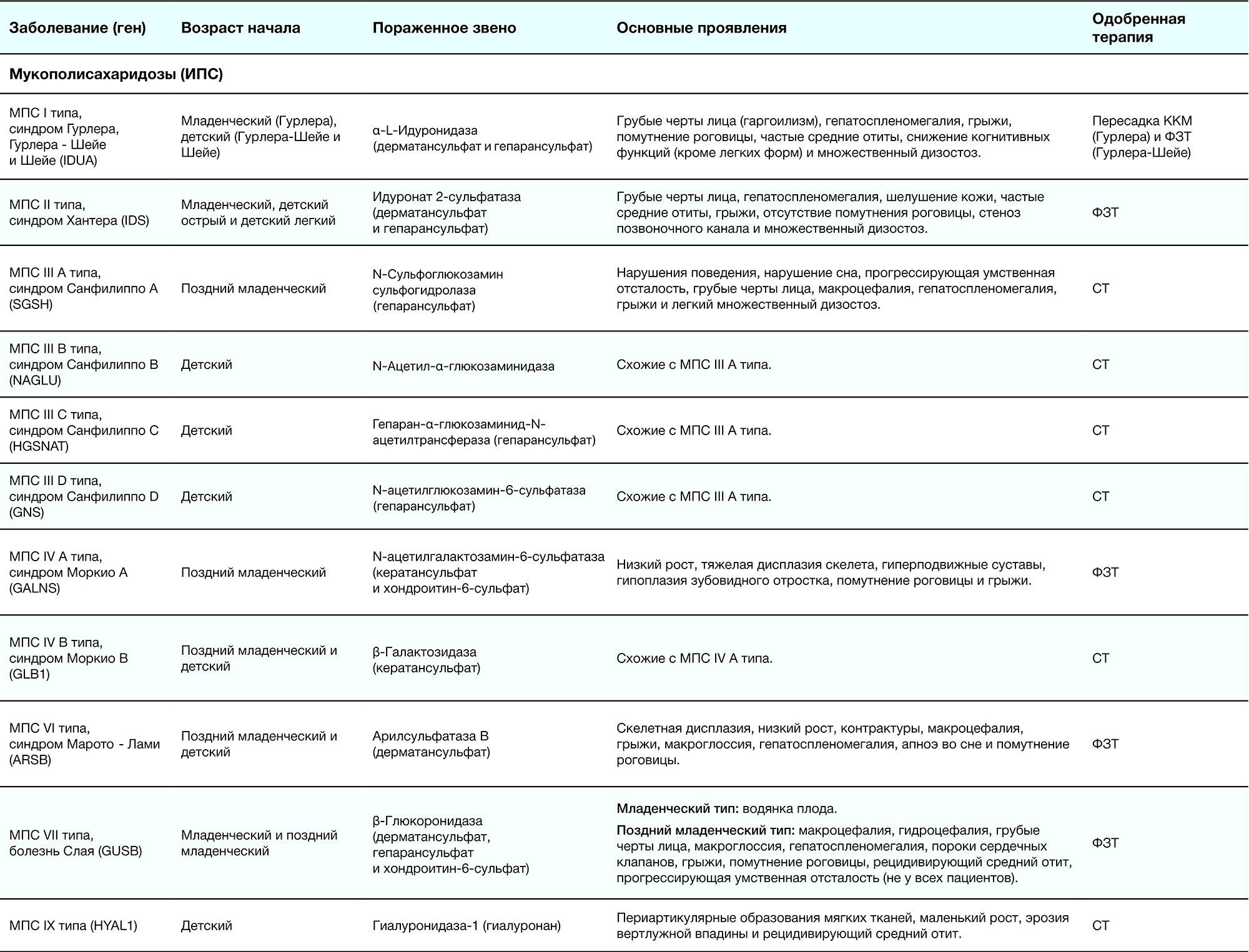
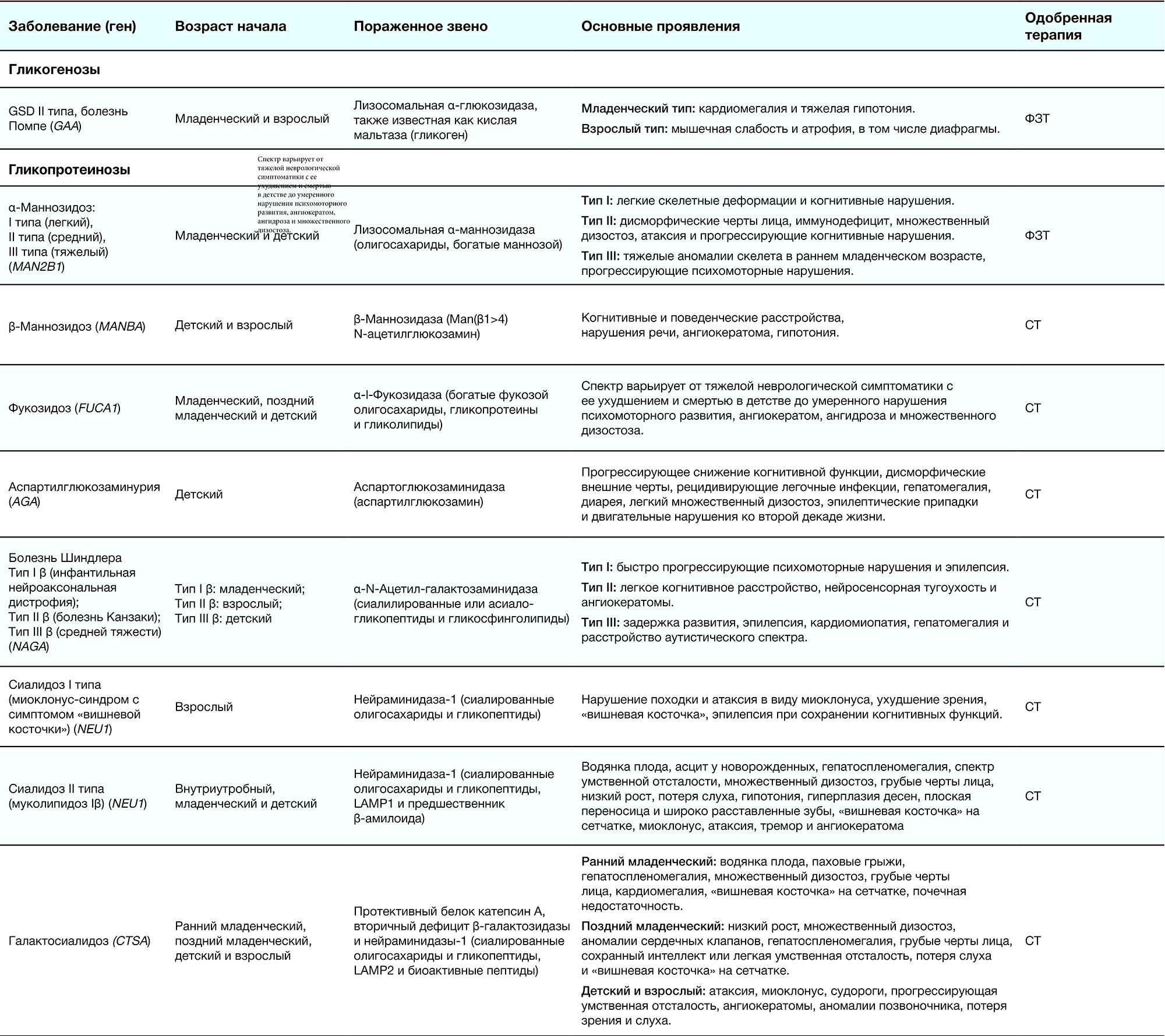
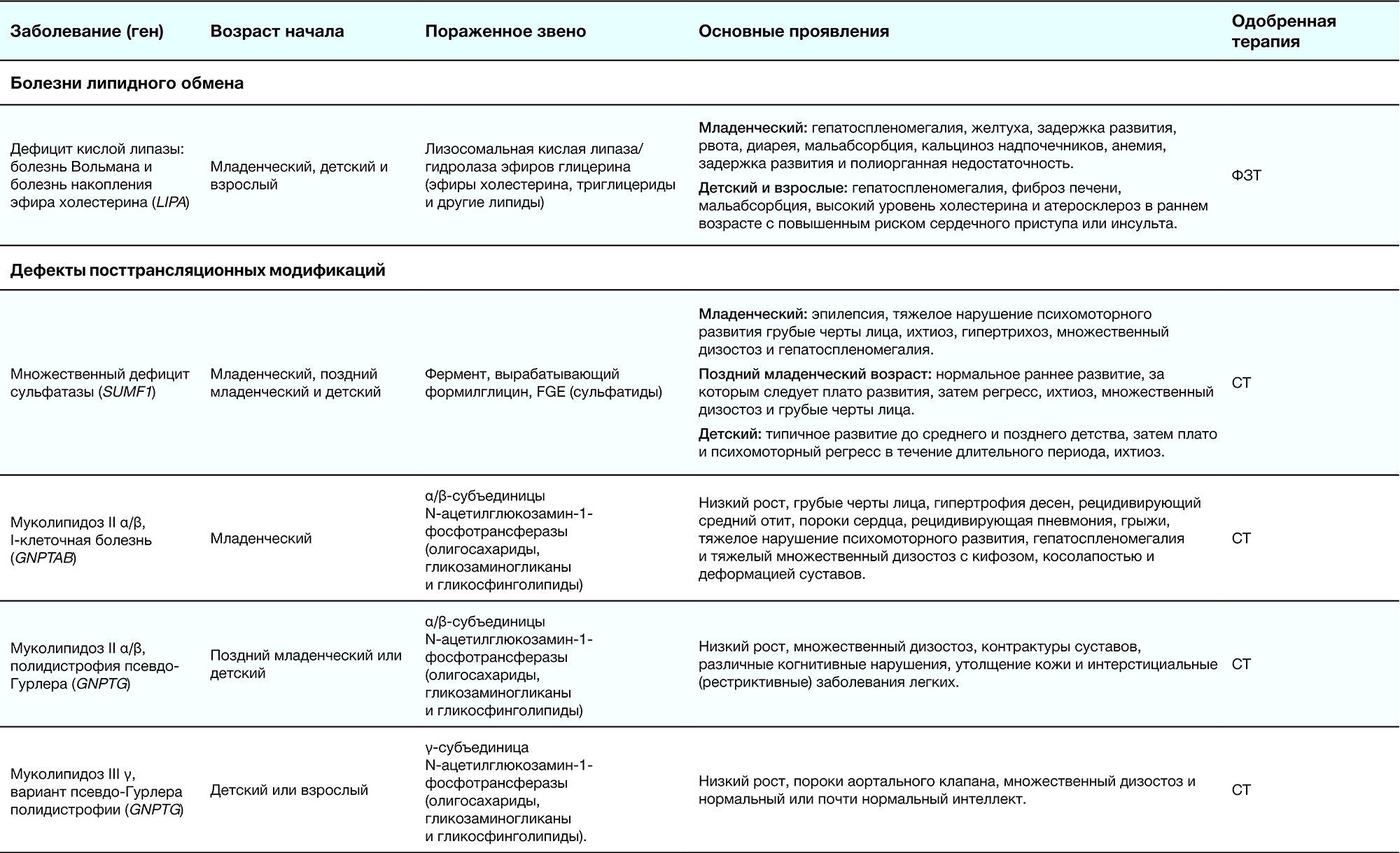
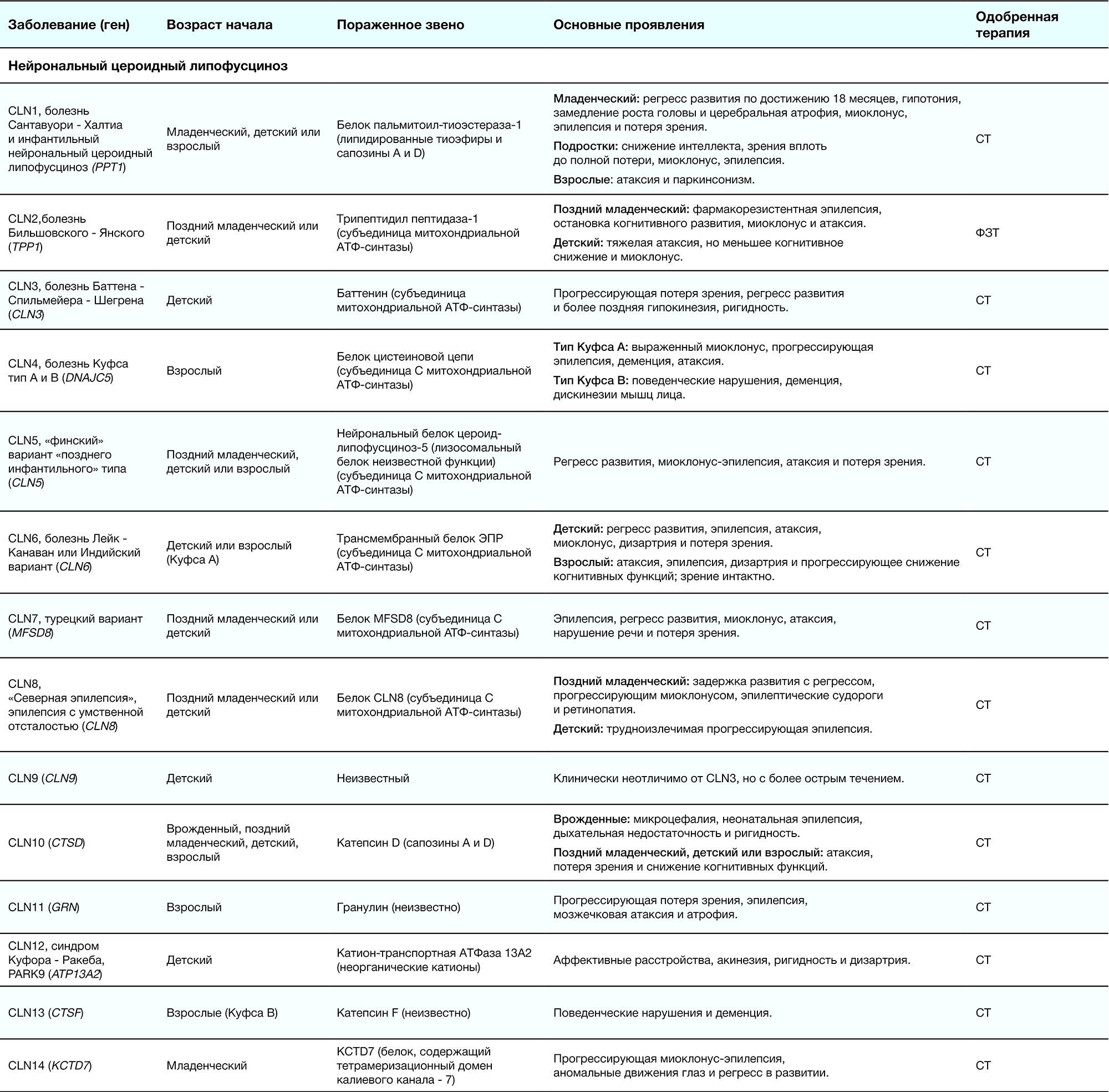
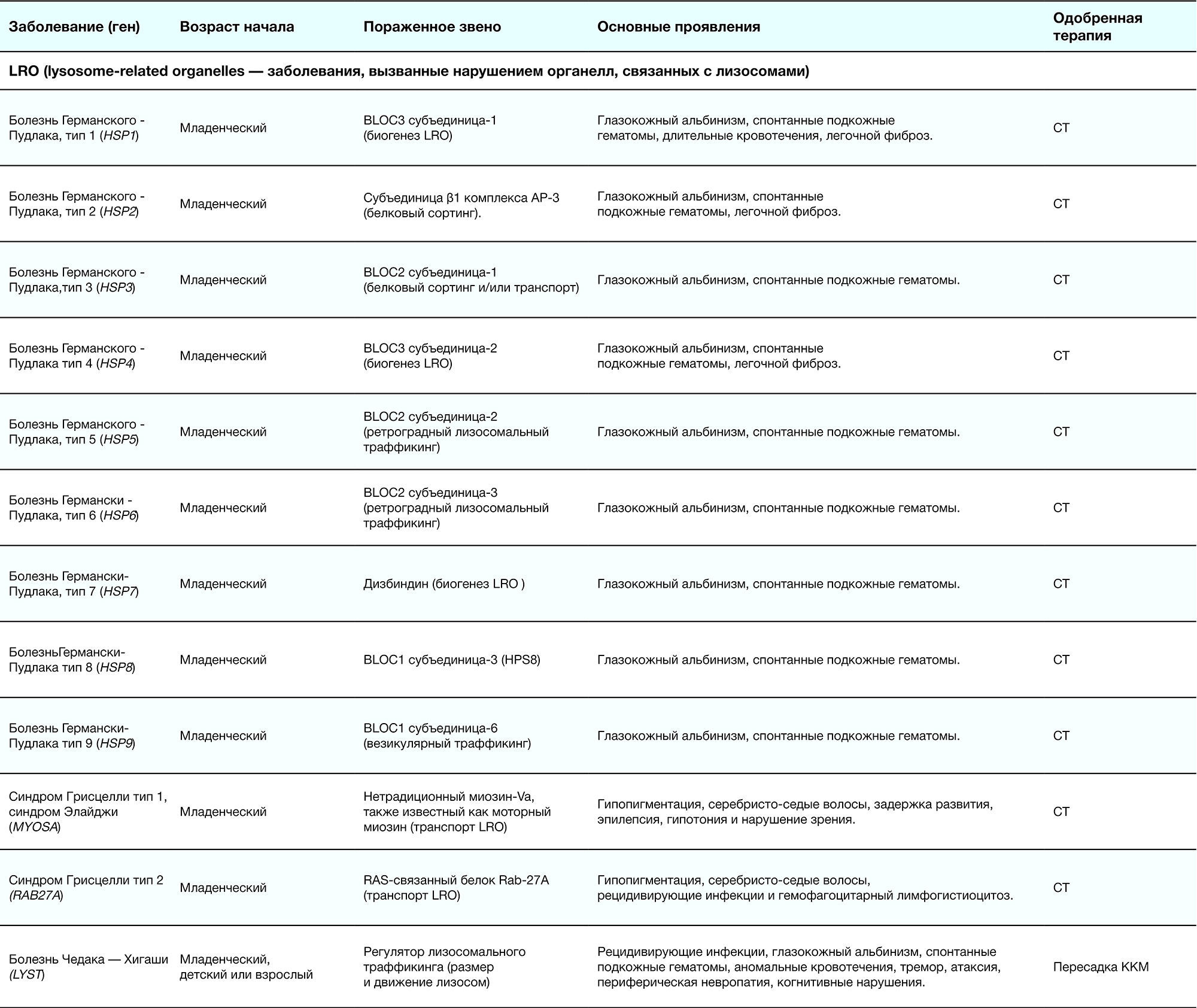
Младенческий возраст 一 в течение первого года жизни с момента рождения; поздний младенческий возраст 一 от года до двух; детский возраст 一 с двух до 16–18 лет; взрослый 一 старше 18 лет.
ФЗТ 一 фермент-заместительная терапия; СРТ 一 субстрат-редукционная терапия; ККМ 一 красный костный мозг; ШТ 一 шапероновая терапия; СТ 一 симптоматическая терапия.
Патогенез
Лизосомы отвечают за распад и переработку макромолекул (включая углеводы, липиды, нуклеиновые кислоты и белки), чем обеспечивают клетку питательными веществами и поддерживают обмен аминокислот и ионов, кальций-зависимых сигналов и соединений. Эти органеллы находятся в динамическом состоянии: они постоянно сливаются с аутофагосомами, фагосомами и плазматической мембраной и играют ключевую роль в межклеточной коммуникации и взаимодействии, иммунном ответе и поддержании клеточного гомеостаза (рис. 1). Кроме того, поздние эндосомы и лизосомы связываются с другими внутриклеточными органеллами (такими как митохондрии и эндоплазматический ретикулум) без слияния с ними, образуя функциональные участки контакта с мембраной. Эти мембранные структуры представляют собой сигнальные микродомены, которые обеспечивают перенос ионов кальция, липидов и других метаболитов между органеллами. Липидный и белковый состав этих контактных участков определяет их функциональные характеристики и влияет на каждую из связанных органелл. Например, сайты контакта митохондрии с лизосомами модулируют деление митохондрий и лизосомную динамику посредством ГТФ-гидролиза Ras-родственного белка Rab7a (RAB7; также известного как RAB7A). Таким образом, понятно, что при ЛБН биохимические свойства и функции этих микродоменов могут быть нарушены из-за изменений в составе эндосомных и лизосомных мембран вследствие измененной активности лизосом.
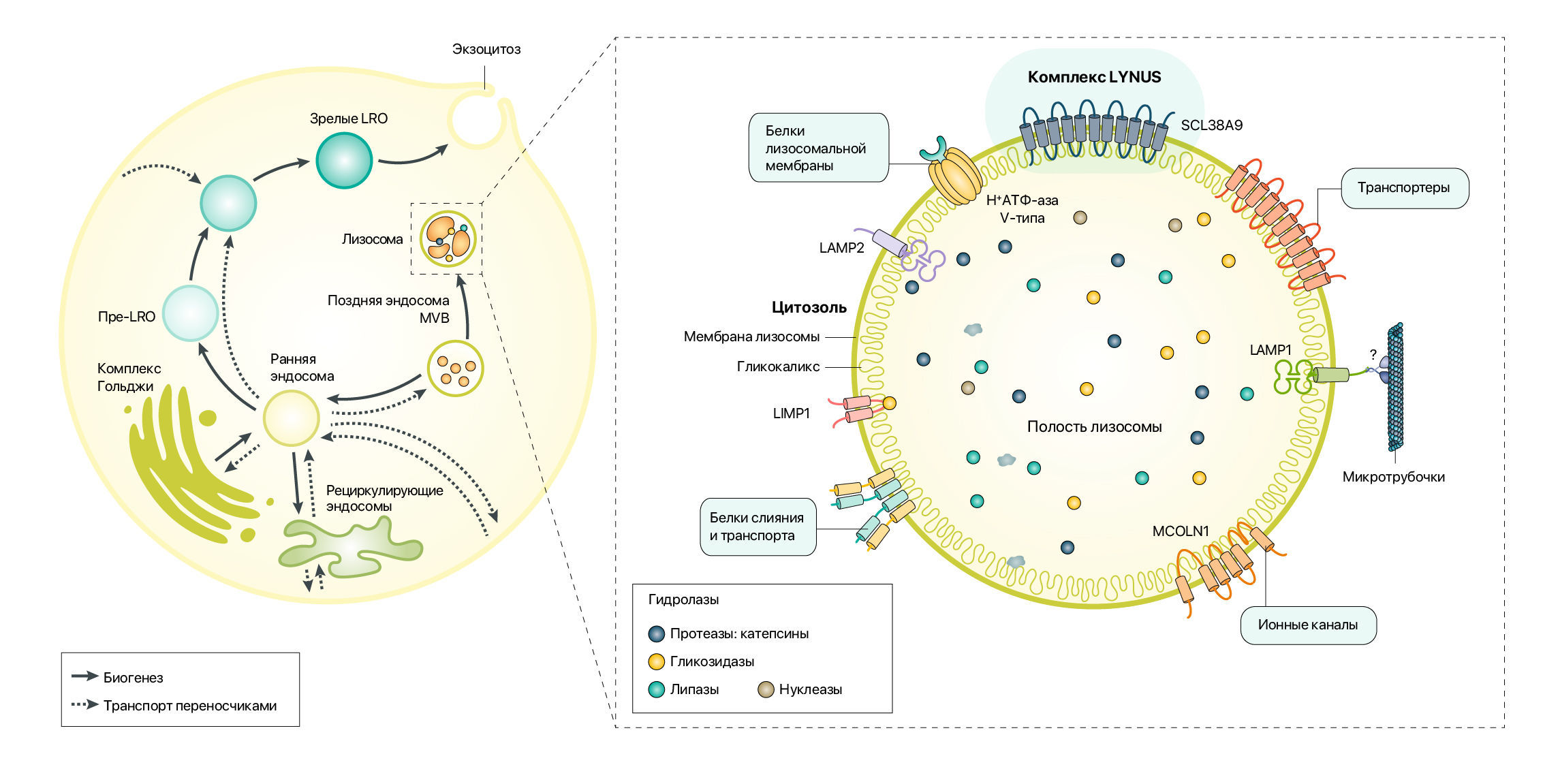
Сплошные стрелки отражают созревание органелл и/или биогенез, а пунктирные стрелки представляют движение макромолекул.
LRO, органелла, связанная с лизосомами; MVB, мультивезикулярное тельце.
Патогенез обусловлен дефектом гена (указаны в табл. 2), что приводит к накоплению метаболитов в самих клетках либо во внеклеточном матриксе. В таблице 2 приведено подразделение на подгруппы дефектов тех ферментов, органелл и т. д., которые и обуславливают развитие заболевания:
- Люминальные белки (любой белок, который хранится в цистернах шероховатого эндоплазматического ретикулума, например, дисульфид изомераза [3]), являющиеся кислотными гидролазами (ферментами) или их активаторами.
- Мембранные интегральные белки [4]:
— Структурные белки (переносчики аминокислот и липидов).
— Ионные каналы, такие как кальциевые каналы.
— Белки траффикинга и слияния.
— Мембранные катаболические ферменты.
— Везикулярная АТФаза, которая действует как протонный насос и сенсор питательных веществ. - Белки, ассоциированные с лизосомами.
Активное участие лизосом в иммунном ответе и клеточной гибели ведет к нарушению последних в случае лизосомных дефектов: избыточное воспаление или недостаточная фагоцитарная активность и, как следствие, иммунный ответ и/или нарушение апоптоза, переход на некроптоз и т. д.
Но нас в первую очередь волнуют неврологические проявления ЛБН.
Пациенты с ЛБН, основным фенотипическим проявлением которых является поражение нервной системы (нейронопатия), часто кажутся нормальными при рождении, однако возраст начала заболевания может значительно варьировать среди разных подтипов расстройств и даже в пределах одного подтипа 一 от тяжелого начала в младенческом возрасте до легких форм, манифестирующих во взрослом периоде. Задержка развития и его регресс происходят вместе с многочисленными неврологическими проявлениями, такими как глазодвигательные нарушения, атаксия, эпи, экстрапирамидные расстройства, спастичность, потеря зрения и нейрокогнитивные расстройства, которые обычно прогрессируют и не поддаются лечению. Разность во времени начала заболевания в основном связана с частичной активностью вовлеченного метаболического пути, потому что даже небольшая остаточная активность может предотвратить накопление метаболитов в большинстве тканей. Однако корреляции между генотипом и фенотипом не всегда четко прослеживаются.
Болезнь Гоше
Болезнь Гоше, наряду болезнью Фабри, является часто встречаемым заболеванием среди сфинголипидозов, имеет аутосомно-рецессивный характер наследования и обусловлено мутацией гена GBA1. Этот ген кодирует фермент глюкоцереброзидазу (GCase), который гидролизует глюкозилцерамид до глюкозы и церамида соответственно.
Исследования историй болезней и популяционно-генетические исследования показали, что пациенты с болезнью Гоше I типа или носители мутаций GBA имеют повышенный риск развития болезни Паркинсона (БП) и деменции с тельцами Леви (ДТЛ). Эти нейродегенеративные состояния характеризуются наличием нерастворимых, олигомерных или фибриллярных включений α-синуклеина в нейронах черной субстанции, гиппокампа и коры головного мозга. Интересно, что обратная взаимосвязь между уровнями глюкоцереброзидазы и α-синуклеина была продемонстрирована экспериментально на клеточных моделях и мышах, но способ взаимодействия между этими молекулами и их вклад в развитие синуклеинопатий полностью не выяснены. Кроме того, глюкозилцерамид напрямую контролирует обратимое изменение α-синуклеина, которое способствует его агрегации и токсичности, открывая возможность терапевтического вмешательства с помощью агентов, снижающих гликосфинголипиды. Необходимы дополнительные исследования, чтобы полностью понять связь между мутациями GBA, особенно у носителей гетерозиготных вариантов, и предрасположенностью к БП. Примечательно, что дисфункция лизосом является связующим звеном между ЛБН и БП (а не конкретно мутациями GBA), поскольку гетерозиготные мутации в 54 из 70 ЛБН чрезмерно представлены у пациентов со спорадической БП.
Клиника
В случае болезни Гоше I типа в рамках поражения нервной системы выделяют легкую сенсорную полинейропатию, которая встречается примерно на 14 % чаще, чем в популяции, и задержку развития. Кроме того, отмечались ранее описанные вторичные формы поражения нервной системы, а именно повышенная вероятность развития БП и ДТЛ, либо на фоне высокой (относительно общей популяции) вероятности развития онкологических заболеваний.
В остальном, в зависимости от степени выработки фермента, возможны асимптоматическое носительство мутации, замедление роста и наступления пубертата, гепатоспленомегалия. Также выделяют эпизоды выраженной костной боли, связанной с аваскулярным некрозом костей, остеомаляцию и остеопороз, костные деформации, инфильтрацию легких и почек клетками Гоше с развитием фиброза легких и почечного повреждения соответственно.
Болезнь Гоше II типа манифестирует в младенческом возрасте (от трех до шести месяцев) мириадой различных неврологических симптомов, в результате чего пациенты доживают примерно до 25 месяцев. Летальная форма встречается в 1 % случаев этой болезни, смерть наступает либо до, либо вскоре после родов [5].
Заболевание начинается с гепатоспленомегалии, последующее развитие симптомов описано триадой: опистотонус, бульбарные расстройства (в частности, тяжелые нарушения глотания) и глазодвигательный паралич. Затем развивается генерализованная миоклонус-эпилепсия, резистентная к противосудорожной терапии, тромбоцитопения, тяжелые поражения легких на фоне аспирации желудочного содержимого и центрального апноэ. Чаще всего смерть наступает в результате дыхательных нарушений.
Внутриутробная форма является самым редким типом болезни Гоше, проявляясь водянкой плода, гепатоспленомегалией, ихтиозом, артрогрипозом, лицевым дисформизом и тромбоцитопенией плода. Смерть наступает антенатально или сразу после рождения.
Болезнь Гоше III типа, которая также зовется юношеской или подострой неврологической формой (от 5 % до 33 % случаев Гоше в некоторых когортах), демонстрирует схожие с болезнью Гоше I типа висцеральные проявления, обычно в сочетании с глазодвигательными расстройствами, проявляющимися обычно до 20 лет. Фенотипы юношеской формы очень разнородны, особенно в отношении неврологических нарушений. Некоторые пациенты демонстрируют только горизонтальный парез взора, как единственное неврологическое проявление, тогда как другие имеют более тяжелые формы с различными неврологическими признаками, включая прогрессирующую миоклонус-эпилепсию (16 % пациентов), мозжечковую атаксию или спастичность (20–50 % пациентов), а иногда и когнитивные, и поведенческие расстройства. Неврологические признаки могут появиться через несколько лет после возникновения проявлений поражения внутренних органов, даже среди пациентов, у которых изначально подразумевалась болезнь Гоше I типа. Заболевание чаще возникает у детей раннего возраста, в половине случаев неврологические симптомы появляются в возрасте до двух лет. Повышается вероятность наступления внезапной смерти. Иногда пациентам может потребоваться оперативное вмешательство в случае тяжелого кифоза, прогрессирующего, несмотря на специфическое лечение. О поражении сердца (с кальцификацией клапана), поражении роговицы и гидроцефалии сообщается в основном среди носителей генотипа c.1342G>C (D409H).
Болезнь Гоше III типа разделяют на четыре подтипа [6]:
- IIIa подтип: легкие висцеральные проявления, но тяжелая и быстропрогрессирующая неврологическая симптоматика, включающая окуломоторную апраксию, мозжечковую атаксию, спастичность, прогрессирующую миоклонус-эпилепсию, не поддающуюся терапии, и деменцию. Прогноз зависит от возраста начала, смерть наступает обычно до 20 лет.
- IIIb подтип: тяжелые висцеральные (гепатоспленомегалия, остеопения, остеонекроз, литические поражения костей, костные кризы [тяжелейшие боли в костях], замедление роста), но легкие неврологические проявления (в большинстве случаев, горизонтальный парез взора).
- IIIc подтип: висцеральные проявления 一 клапанные пороки сердца, восходящей части аорты, легкая гепатоспленомегалия, помутнение роговицы, костные поражения и аномалии; неврологические 一 надъядерный парез взора, гидроцефалия.
- Норрботтенский тип характеризуется ранним началом, массивным поражением внутренних органов, прогрессирующим кифосколиозом и умеренными когнитивными нарушениями. Географически Норботтен расположен на севере Швеции, где, вероятно, и был описан этот подтип. Эта форма встречается примерно в 40 % всех известных случаев болезни Гоше в Швеции. Это объясняется эффектом основателя (о чем уже писалось выше). При классической форме заболевания ранняя манифестация и клинические особенности могут ошибочно привести к постановке диагноза болезни Гоше I типа. Первые клинические симптомы появляются в среднем в возрасте одного года. Наблюдаются висцеральные поражения (гепатоспленомегалия, часто требующая спленэктомии в раннем возрасте, гематологические расстройства [тромбоцитопения, анемия], скелетные аномалии [в том числе кифосколиоз] и инфильтраты сетчатки) и неврологические симптомы (супрануклеарный парез горизонтального взора, страбизм, атаксия, легкая спастичность в ногах, миоклонус-эпилепсия или фокальная с нарушением осознанности, а также медленно прогрессирующее когнитивное снижение, ведущее к деменции).
Иные формы болезни Гоше
Дефицит сапозина С
Сапозины являются важными кофакторами на нескольких стадиях лизосомной деградации сфинголипидов, которые включают семейство из четырех небольших гликопротеинов, известных как сапозины A, B, C и D. Эти гомологичные белки получены в результате последовательного протеолиза общего предшественника белка просапозина (PSAP), кодируемого геном PSAP на 10 хромосоме. Сапозин С является активатором гидролитической активности GCase. Он также играет защитную роль в протеолитической деградации GCase. В результате мутация в домене сапозина С в PSAP приводит к дефициту первого, что является очень редкой причиной болезни Гоше с нормальной активностью GCase, обычно проявляющейся как болезнь Гоше III типа.
Также отмечаются промежуточные формы II–III типов. Проще говоря, промежуточные формы предполагаются среди пациентов, проживших более двух лет и имеющих смешанную симптоматику II и III типов.
GM1-ганглиозидоз [7]
GM1-ганглиозидоз дифференцируется на три типа и в каждом случае на передний план выходит неврологическая симптоматика.
- 1 тип GM1-ганглиозидоза манифестирует примерно в возрасте шести месяцев (до 12 месяцев), смерть наступает до двух лет в результате аспирационной пневмонии либо сердечной недостаточности на фоне развившейся рестриктивной кардиомиопатии [8]. Пациенты могут иметь грубые черты лица (гаргоилизм), скелетную дисплазию, гепатоспленомегалию, симптом «вишневой косточки» на сетчатке, слепоту, нейросенсорную тугоухость, психомоторный регресс, мышечную гипотонию c последующей эволюцией в генерализованную спастистичность и гиперрефлексию, эпилепсию, гиперэкплексию и множественные стигмы дизэмбриогенеза.
- GM1 2-го типа подразделяют на поздний младенческий (возраст начала один — три года) и детский (начинается в 3 — 10 лет) фенотипы. Проявляется прогрессирующим снижением когнитивных функций, нарушением моторики и речи, также может наблюдаться легкое помутнение роговицы, гепатоспленомегалия и/или кардиомиопатия. Типичное течение GM1-ганглиозидоза 2-го типа характеризуется прогрессирующим неврологическим дефицитом, костными дисплазиями среди некоторых пациентов (включая кифоз и аваскулярный некроз головки бедренной кости) и прогрессирующими затруднениями с кормлением, ведущими к риску аспирации в виду бульбарной симптоматики. Смерть наступает обычно до 20 лет.
- GM1-ганглиозидоз 3-го типа интересен хроническим прогрессирующим течением, маскирующимся под личинами других нейродегенеративных заболеваний. Манифестация заболевания обычно происходит между подростковым периодом и третьей декадой жизни. Заболевание может проявить себя в виде медленно прогрессирующего снижения когнитивных функций (вплоть до деменции) с паркинсонизмом и экстрапирамидными нарушениями. На первое место может выйти генерализованная дистония (обычно так и бывает), речевые расстройства и акинетико-ригидный паркинсонизм, а когнитивные нарушения возникнут позднее в течение жизни. Также сопутствуют скелетные аномалии (обычно легкий кифосколиоз, низкий рост) и кардиомиопатия.
Клинический случай GM1-ганглиозидоза 3-го типа [9]
Мужчина 23 лет, родился в результате кровного брака в срок, рос и развивался без особенностей, никто из членов семьи не болен. Его родители заметили, что к четырем годам пациент перешел на ходьбу на носках, с целью коррекции была подобрана специальная обувь (без эффекта). Во время учебы в школе с каждым годом ухудшалась его успеваемость, и он не смог закончить восьмой класс. Ему пришлось прекратить обучение в обычной школе, но в конце концов удалось сдать экзамены в средней школе в частном порядке. В возрасте 16 лет его родители наблюдали вычурные движения лица, которые усиливались, когда он говорил, речь постепенно ухудшалась. Было отмечено, что в этот период у него постепенно изменялась обычная поза постановки рук и ног. Терапия леводопой-карбидопой в максимальной суточной дозировке до 300 мг в какой-то степени улучшила моторные функции, однако ее пришлось отменить, поскольку у мужчины развился психоз, требующий ЭСТ и антипсихотиков. Была проведена правосторонняя стереотаксическая таламотомия, которая уменьшила контралатеральную ригидность примерно на 50 %.
У пациента не отмечалось гепатоспленомегалии, помутнения роговицы или катаракты, при осмотре глазного дна патологии не выявлено, глазодвигательные функции в норме. Отмечалась выраженная лицевая дистония с медленными извилистыми движениями, вовлекающими лобную мышцу, круговую мышцу рта, губы и углы рта (рис. 2), также присутствовали дистония с брадикинезией рук. Эти особенности свидетельствовали о генерализованной дистонии с паркинсонизмом. Обычные рентгеновские снимки кистей рук и грудопоясничного отдела позвоночника не выявили отклонений. Речь и дистония рук пациента улучшились на фоне терапии высокими дозами тригексифенидила (24 мг/день), но и это лечение пришлось прекратить из-за нарастающей раздражительности. В настоящее время пациенту 29 лет, он находится под амбулаторным наблюдением с минимальным прогрессированием неврологического дефицита. Исследование костного мозга показало наличие нескольких крупных пенистых клеток с обильной цитоплазмой и маленьким, круглым, везикулярным, эксцентрично расположенным ядром. В цитоплазме были обнаружены множественные короткие эозинофильные полосы, которые придавали клеткам характерный вид «сморщенной папиросной бумаги», напоминающий клетки Гоше.

Рисунок 2. Пациент с GM1-ганглиозидозом из клинического случая [9]
GM2-ганглиозидоз
GM2-ганглиозидоз представляет группу из трех заболеваний: Тея — Сакса, Сандхоффа и AB вариант. Заболевания эти манифестируют в младенческом, позднем младенческом или раннем детском, позднем детском и взрослом периодах соответственно, однако это правило выполняется не всегда.
Болезнь Тея — Сакса [10]
Болезнь Тея — Сакса обусловлена мутацией гена, кодирующего субъединицу а гексозаминидазы А, что приводит к значительному снижению концентрации этого вещества. Чаще протекает как острое заболевание, проявляясь в младенческом возрасте в виде рефрактерной эпилепсии, аксиальной гипотонии, симптома “красной вишневой косточки» на сетчатке, психомоторного регресса, гиперэкплексии. Даже в случае лучшего ухода за больным, смерть наступает в возрасте четырех-пяти лет из-за рекуррентных инфекций. Поздняя манифестация встречается значительно реже и представлена двигательными нарушениями, атаксией, психотическими нарушениями, часто не чувствительными к терапии, нейродегенерацией. Один из таких клинических случаев представлен ниже.
Необычный клинический случай болезни Тея — Сакса [11]
Женщина 53 лет, с детства беспокоили крампи в ногах и трудности при беге, но в остальном она была здорова. С 20 лет появились затруднения при вставании со стула. С 28 лет при подъеме по лестнице, развилась широкая походка, что послужило поводом для ее первого неврологического обследования. По происхождению она ашкенази, анамнез не скомпрометирован по двигательным нарушениям, родители не находятся в родстве. Ранее в записях ее обследования в возрасте 28 лет сообщалось о симметричной проксимальной слабости рук и ног. По старым данным на ЭНМГ описывалась «хроническая нейропатия», но дальнейшие подробности отсутствовали. Биопсия мышц выявила неспецифические изменения, в т. ч. преобладание волокон типа I с повышенной окислительной активностью в субсарколеммальной области во многих волокнах и небольшое увеличение межпучковой соединительной ткани. Сообщалось, что МРТ головного мозга на тот момент без особенностей.
Примерно в возрасте 35 лет подниматься по лестнице и вставать со стула стало еще труднее, присоединились частые падения. Миалгий не было, мышечной атрофии не выявлено. ЭНМГ на то время указывала на болезнь двигательных нейронов (по записям), в результате чего был предложен диагноз спинальной мышечной атрофии. Генетическое тестирование не проводилось.
После 40 лет присоединился периодический тремор рук и ухудшилась мелкая моторика, что повлияло на ее почерк. Речь стала невнятной. Проблемы с равновесием приводили к падению и травмам, а к 51 году была вынуждена использовать ходунки.
В 52 года была повторно обследована неврологом. На МРТ головного мозга выявлена изолированная выраженная атрофия мозжечка (рис. 3), а на ЭНМГ отмечалась хроническая аксональная моторная полинейропатия. Генетические исследования не выявили мутаций ни в генах, кодирующих СЦА 1, 2, 3, 5, 6, 7, 8, 10, 12, 13, 14, 17, 28 или дентатопаллидольюисову атрофию, ни в генах FXN, APTX, SETX, POLG1, SIL1 или TTPA.
К 53 годам у пациентки были сохранны когнитивные функции, речь беглая, монотонная. Глазодвигательных или зрительных нарушений обнаружено не было. Мышечная сила с трехглавой, подвздошно-поясничной и четырехглавой мышц была оценена как 4,5 с обеих сторон, в остальном — 5 баллов, тонус физиологический. Рефлексы 3+ в руках и 1+ в ногах без патологических знаков. Присутствовал симптом Говерса. Пациентка имела интенционный тремор в руках, атактическую походку, неспособность совершать тандемную ходьбу, передвигалась с опорой на ходунки, иногда выявлялся хореоатетоз. Поверхностная и глубокая чувствительность без нарушений.
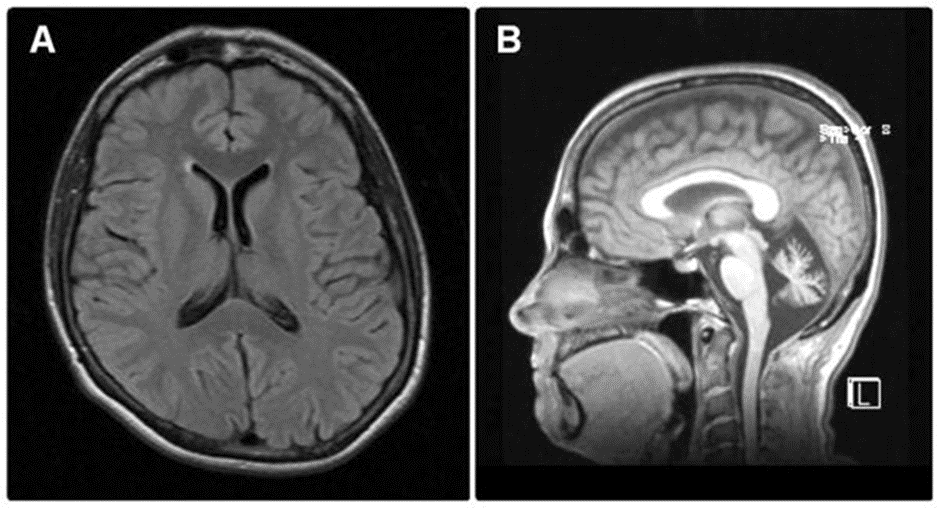
Рисунок 3. Нейровизуализация (МРТ) пациентки с болезнью Тея — Сакса [11]
Уровни витамина Е и функции щитовидной железы были в норме. Общее количественное определение гексозаминидазы было нормальным (13,6 Ед/л, референсные значения 10,4–23,8 Ед/л), в то время как активность гексозаминидазы А отсутствовала. Генетическое тестирование выявило мутации TATCins1278 и Gly269Ser.
Болезнь Сандхоффа
Болезнь Сандхоффа обусловлена дефектом гена, кодирующего b субъединицу глюкозооксидазы A. Характерно подострое течение заболевания, проявляющееся в среднем в возрасте от двух до девяти месяцев. Заболевание проявляется атаксией, миоклонусом, регрессией двигательных навыков или прогрессирующей моторной слабостью, психотическими эпизодами, умственной отсталостью, дистонией, эпилепсией, гиперэкплексией, слуховыми и зрительными нарушениями. При классической форме, как и в случае болезни Тея — Сакса, прогноз неутешительный, смерть наступает обычно в возрасте до трех лет. Однако возможны и исключения в виде поздней манифестации, включающей широкий спектр синдромов, в том числе спиноцеребеллярную атаксию, сенсорно-моторную полинейропатию, болезни двигательного нейрона, тремор, дистонию и психотические эпизоды (клинический кейс ниже).
Клинический случай взрослой формы болезни Сандхоффа [12]
Женщина 40 лет была направлена на обследование для оценки медленно прогрессирующей в течение трех лет слабости нижних конечностей. Она сообщила о слабости обеих ног, трудностях при вставании со стула, плохом равновесии и подергивании мышц бедра. Дополнительные симптомы включали беспокойство, колебания настроения, снижение концентрации, общую усталость и плохой сон. Пациентка отрицала онемение, боль, слабость в руках, затрудненное дыхание, нарушения речи и глотания.
При медицинском осмотре у нее была легкая слабость в сгибателях бедра, сгибателях и разгибателях голени, с сохранной силой во всех других группах мышц. Отмечалась легкая атрофия разгибателей голени с редкими фасцикуляциями. Глубокие сухожильные рефлексы в норме, за исключением коленного, который был снижен. Тандемная походка незначительно нарушена. В остальном неврологическое обследование в пределах нормы.
На МРТ головного мозга отмечалась легкая атрофия головного мозга и мозжечка с преимущественным поражением верхней части червя. МРТ поясничного отдела позвоночника демонстрировала двусторонний стеноз выхода спинномозговых корешков на уровнях L5–S1. ЭМГ в пределах нормы, за исключением нейрогенного характера поражения в разгибателях голени и сгибателях бедра. Уровень КФК в норме.
Двигательная симптоматика и результаты ЭМГ подразумевали хроническую форму заболевания нижнего мотонейрона, но результаты МРТ не объясняли симптомы. Картина не была типичной ни для одного из известных заболеваний нижних мотонейронов. Поскольку не существует всеобъемлющей панели для генетических заболеваний двигательных нейронов, и пораженный ген не может быть точно определен клинически, для дальнейшей оценки было выполнено полноэкзомное секвенирование (whole exome sequencing, WES). WES выявило сложную гетерозиготную мутацию в гене HEXB на хромосоме 5 (патогенный вариант c.298delC и вероятный патогенный вариант G473S). Последующее ферментативное тестирование гексозаминидазы выявило снижение общей активности гексозаминидазы в лейкоцитах (14 % от нормального уровня), что соответствует ганглиозидозу GM2. Кроме того, отмечалось высокое соотношение активности а- гексозаминидазы А к активности b-субъединицы (79 %), что подтверждает диагноз болезни Сандхоффа.
AB вариант
Это наиредчайший представитель GM2-ганглиозидозов, обусловленный дефектом гена, кодирующего белок активатор GM2. Всего описано около 10 таких случаев [13]. Клинически заболевание может быть неотличимым от типичного случая болезни Тея — Сакса или же принимать хроническую форму с манифестацией в подростковом или взрослом периодах.
Важным замечанием будет и то, что данные формы могут быть неотличимы друг от друга клинически, проявляясь в виде острого фенотипа в раннем младенческом возрасте, подострым в позднем младенческом возрасте и до 10 лет и хроническим с подросткового возраста.
Болезнь Помпе
Болезнь Помпе дифференцируется на две формы [14]:
- Первая форма — классическая (манифестация до одного года), включающая выраженную мышечную слабость (с бульбарной симптоматикой) и гипотонию, кардиомегалию, гепатомегалию, макроглоссию, задержку развития (вероятно, вследствие трудностей с кормлением), нарушение слуха. Слабость дыхательных мышц приводит к дыхательной недостаточности и частым респираторным инфекциям, что и является основной причиной смертности среди больных.
- Вторая форма — неклассическая (взрослая), проявления которой могут напоминать таковые из классической формы с различной вариабельностью и меньшей степенью выраженности. Основным, а иногда и единственным симптомом может быть проксимальная мышечная слабость. Кардиомиопатия встречается редко. Единственной жалобой больного может быть «неуклюжесть», «неловкость» при выполнении каких-либо упражнений.
В последнее время спектр различных проявлений поздней формы значительно расширился, и в добавок к миопатии и кардиомиопатии наблюдаются синдром ригидного человека, остеопороз, сколиоз, апноэ сна, нейросенсорная тугоухость, офтальмоплегия, нейропатия тонких волокон, интракраниальные аневризмы, расстройства ЖКТ и нарушение тазовых функций [15].
Клинический кейс болезни Помпе [16]
Мужчина 36 лет, поступил в отделение неотложной помощи с жалобами на выраженную одышку, непродуктивный кашель и мышечную слабость. Ввиду развития дыхательной недостаточности второго типа была проведена интубация с последующим переходом на неинвазивную вентиляцию легких через несколько недель после поступления в стационар.
Со слов пациента, он отмечал аксиальную мышечную слабость «сколько себя помнит». Ухудшение состояния началось примерно за три-четыре года до госпитализации в виде прогрессирующего снижения толерантности к физической нагрузке. Два года назад присоединились дыхательные нарушения, вплоть до того, что пациент примерно за три месяца до поступления в больницу не мог спокойно лежать на спине из-за нарастающей одышки. Отмечалось повышение уровней АЛТ и АСТ в течение пяти лет, уровень КФК не исследован. Год назад мужчина обращался к неврологу с тремором, на проведенной нейровизуализации без патологии. Семейный анамнез — без особенностей.
Физикальное обследование выявило атрофию параспинальных мышц, слабость проксимальных отделов ног (до 3 баллов) и гипертрофию икроножных мышц. Эхо-КГ в норме, форсированная жизненная емкость легких (ФЖЕЛ) составляла 30 % с падением до 12 %, в положении на спине; тест с 6-минутной ходьбой — 370 метров.
КФК 1810 Ед/л, тетрасахариды в моче до 110 (норма < 20 ммоль/моль креатинина). По анализу сухой капли крови выявлено снижение функции кислой альфа-глюкозидазы до 0,3 мкмоль/литр/час (норма 2,4–10,2). Мышечная биопсия показала вакуолизированные мышечные волокна и включения гликогена. Генетическое исследование подтвердило болезнь Помпе. Была начата ФЗТ, что позволило замедлить прогрессирование заболевания, однако пациенту все равно требовалась неинвазивная вентиляция легких во время сна.
Болезнь Фабри
Болезнь Фабри обусловлена X-сцепленной мутацией гена, кодирующего α-галактозидазу А, при которой нарушается образование соответствующего фермента. Заболевание варьирует по своим проявлениям, подразделяется на классическую форму, проявляющуюся в детском возрасте (приблизительно от 3 до 10 лет, причем у девочек оно манифестирует позже), и позднюю форму, дающую о себе знать в возрасте от 40 до 70 лет. В зависимости от выработки α-галактозидазы А заболевание имеет разные фенотипы, характеризующиеся как мультиорганными поражениями (почек, сердца, нервной системы, глаз и кожи), так и поражением одной системы органов. В целом, продолжительность жизни снижена у мужчин на 15 лет, у женщин — на пять лет [17].
При классической форме болезни Фабри первые симптомы, включая хроническую нейропатическую боль и эпизодические сильные болевые кризы, обычно возникают в детстве. Другие частые ранние проявления — гипогидроз, ангиокератомы, расстройства ЖКТ (вздутие живота, диарея, боль в животе) и характерная вихревая кератопатия. Чуть позже может присоединиться альбуминурия и гломерулосклероз с последующим развитием хронической болезни почек (ХБП) во взрослом периоде. Прогрессирование ХБП, гипертрофия левого желудочка, нарушение ритма сердца, потеря слуха, транзиторные ишемические атаки, ишемические инсульты свойственны раннему взрослому периоду. Накопление GL-3 в тканях миокарда, а также воспалительные и нейрогормональные механизмы, приводящие к кардиальной клеточной и сосудистой дисфункции, вероятно, вносят вклад в проявления поражения сердца. Сообщалось также о легочных проявлениях (например, одышка, хрипы, сухой кашель). У пациентов с более поздним фенотипом типичны кардиологические проявления (например, гипертрофия левого желудочка, аритмия) и, в редких случаях, ХБП, наблюдающиеся с четвертой по седьмую декаду жизни, отражая отсроченное начало и более медленное прогрессирование заболевания. Спектр заболевания у гетерозиготных пациентов женского пола варьируется от бессимптомного или легкого, с более поздними проявлениями фенотипа, которые обычно поражают только один или несколько органов, до тяжелых проявлений (как у пациентов мужского пола с фенотипом классического заболевания).
Таблица 3. Клиническая характеристика болезни Фабри [17]
Клинический случай болезни Фабри [18]
Юноша 17 лет, с раннего детства страдал гипогидрозом. При исследовании кожи отмечались генерализованные телеангиэктазии, включая ладони, подошвы и слизистые оболочки, появившиеся за последние 7 лет (рисунок A–C). Кроме того, пациент жаловался на парестезии дистальных отделов рук и ног, беспокоящие 13 лет. Молодой человек невысокого роста, с задержкой полового развития. Его дядя по материнской линии также жаловался на гипогидроз и боль в руках и ногах; а скончался в возрасте до 40 лет от некоего сердечно-сосудистого заболевания (не уточняется). Биопсия кожных образований соответствовала ангиокератоме. Активность фермента α-галактозидазы А ниже нормы. На ЭНМГ отмечалось нарушение проводимости по нервным волокнам. Был выявлен остеопороз (Z-критерий «–4»). Функция щитовидной железы и концентрация тестостерона в норме. Сердечные и легочные функциональные тесты и офтальмологические исследования — без особенностей. МРТ головного мозга и УЗИ брюшной полости в норме.
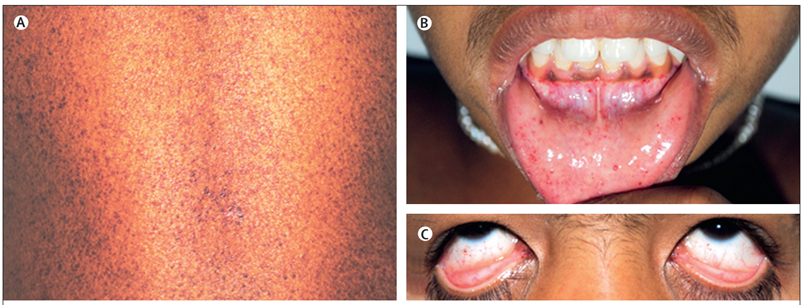
Рисунок 4. Телеангиэктазии пациента из клинкейса [18]
Метахроматическая лейкодистрофия (МЛД) [19]
МЛД характеризуется повреждением миелиновой оболочки, покрывающей большинство нервных волокон ЦНС и ПНС, что приводит к прогрессирующим моторным и когнитивным нарушениям в качестве клинических проявлений.
МЛД получила свое название из-за наличия в пораженных клетках метахроматических гранул, образовавшихся в результате накопления сульфатидов и сфинголипидов, представленных в миелине. При МЛД сульфатиды накапливаются в олигодендроцитах, микроглии, некоторых нейронах ЦНС, шванновских клетках и макрофагах ПНС. Они также накапливаются в клетках внутренних органов, например, в желчном пузыре, что увеличивает риск злокачественных новообразований этого органа. МЛД вызывает демиелинизацию, что приводит к нарушению двигательной функции, спастическому тетрапарезу, атаксии, атрофии зрительного нерва и когнитивным нарушениям.
Заболевание дифференцируется на три формы, от более тяжёлого к менее:
- Инфантильная форма (новорожденных) проявляется до 30 месяцев жизни. Практически полное отсутствие минимальной активности фермента ARSA обуславливает тяжелую нейродегенерацию. В спектр симптомов входят психомоторные нарушения и регресс развития, тетрапарез, атаксия, гиперкинезы, эпилептические припадки, полинейропатия, бульбарные расстройства, нарушения слуха и зрения. При манифестации в позднем младенчестве пациент доживает до подросткового возраста.
- Ювенильная (или детская) — от 3 до 16 лет. Характерны когнитивные и поведенческие расстройства, а также двигательные и сенсорные нарушения, обусловленные поражением как центральных, так и периферических нервных волокон. Также возможны эпилептические припадки.
- Взрослая форма наблюдается среди лиц старше 16 лет и проявляется когнитивными и поведенческими нарушениями, атаксией, полинейропатией, эпилептическими приступами. Пациенты испытывают депрессивные расстройства и резкие перепады настроения. Другой типичный признак — психотические симптомы, такие как галлюцинации и иллюзии, которые могут быть связаны с нарушением межкортикальных и кортико-подкорковых связей, особенно с вовлечением лобных долей. МЛД у взрослых часто ошибочно диагностируют как раннюю деменцию или шизофрению из-за медленного прогрессирования, а также периодов относительной стабильности и частичной регрессии клиники. Заключительная стадия заболевания похожа на позднюю инфантильную и ювенильную формы. Ожидаемая продолжительность жизни пациента составляет около 20–30 лет после постановки диагноза.
Клинический случай метахроматической лейкодистрофии [20]
Женщина, 41 год, врач-кардиолог. Обратилась в офтальмологическую клинику в июле 2014 года с жалобами на бинокулярную нечеткость зрения и боль при движении глаз в течение трех месяцев. Острота зрения у пациентки продолжала снижаться, что сопровождалось ощущением отечности глазных яблок и болью. Боль была периодической, продолжительностью около двух часов за эпизод, усиливалась в период активности и уменьшалась после отдыха. Бинокулярное зрение пациентки ухудшилось, однако поля зрения, цветовое восприятие и результаты осмотра глазного дна при обращении были в пределах нормы. Ранее она обращалась в другую больницу с жалобами на сниженное настроение, после чего ей был выставлен диагноз депрессии. Несмотря на лечение антидепрессантами, симптомы продолжали прогрессировать. Кроме того, ее муж вспомнил, что полгода назад, когда она готовилась к экзамену на повышение квалификации, она отмечала ухудшение памяти, обучаемости и концентрации внимания. Ввиду ухудшения зрения и психомоторных нарушений женщина была направлена в неврологическое отделение. Семейный анамнез без особенностей. При поступлении была ориентирована в личности, месте, времени. Отмечалась брадифрения, дезорганизация поведения. Шкалы MOCA и MMSE — 16 и 21 балл соответственно. Других симптомов при неврологическом обследовании не выявлено.
Исследование на антитела к ANA, ACA, ANCA, TGAB, ATPO и dsDNA — в пределах нормы. Результаты рутинного тестирования на сифилис и ВИЧ были отрицательными. Кривые зрительных вызванных потенциалов (ЗВП) (N75, P100 и N145) не обнаруживались (рис. 4). ЭЭГ продемонстрировало аномальное двустороннее синхронное замедление. Уровень белка в ЦСЖ был повышен (1,094 г/л), но IgG и лейкоциты — в пределах нормы. МРТ головного мозга показала диффузную гиперинтенсивность белого вещества в области полуовальных центров и лучистых венцов с двух сторон на T2 и T2-FLAIR, демонстрируя характер демиелинизации по «тигроидному» типу или по типу «кожи леопарда». МРТ шейного и грудного отделов в норме (рис. 6).
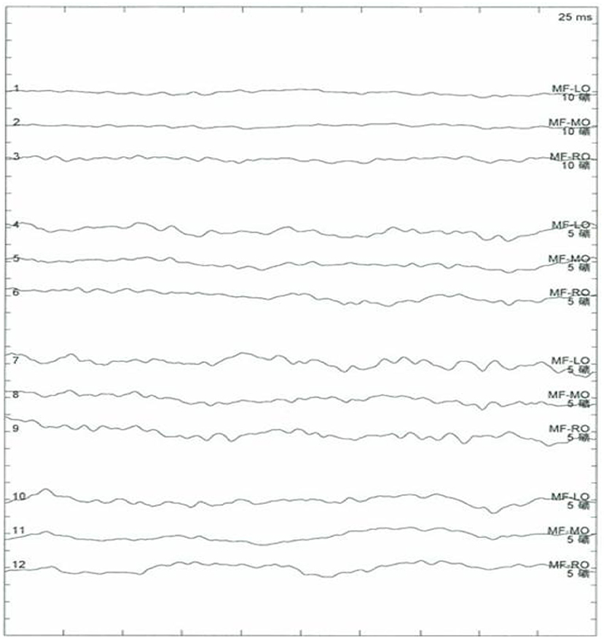
Рисунок 5. Зрительные вызванные потенциалы пациентки не фиксируются [20]
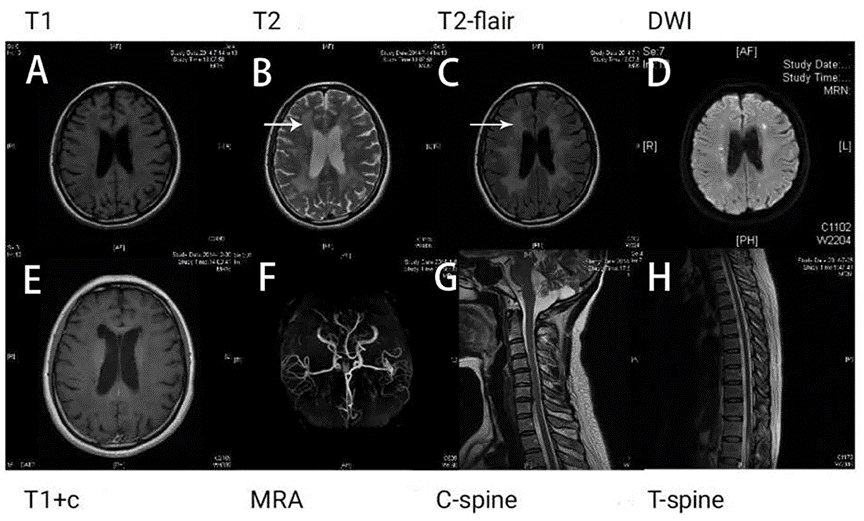
Рисунок 6. Поражение белого вещества пациентки из клинического случая [20]
«Тигроид» или «шкура леопарда» присутствуют на Т2 и T2-FLAIR (В и С, белая стрелка). Гипоинтенсивные очаги на T1 (A) и гиперинтенсивные на последовательности DWI (D). При введении контраста усиления сигнала не наблюдается (E). МР-ангиография (F), а также визуализация шейного (G) и грудного отделов спинного мозга (H) без патологии.
На основании результатов клинических и лабораторных исследований был рассмотрен диагноз рассеянного склероза, начато лечение пульс-терапией метилпреднизолоном (1000 мг/сут в течение 5 дней). После лечения пациентка сообщила о легком улучшении остроты зрения, но результаты офтальмологического обследования не изменились. Повторное исследование ЦСЖ выявило повышенный уровень белка (1,005 г/л) и нормальный индекс IgG (0,611).
Через пять месяцев состояние пациентки продолжало медленно прогрессировать, и она обратилась в другую больницу. Был рассмотрен тот же диагноз рассеянного склероза, и на этот раз был начат плазмаферез. Пациентка отметила незначительное улучшение зрения, но через 20 дней у пациентки появилась слабость в нижних конечностях, ввиду которой она не могла ходить самостоятельно. В декабре 2014 года ее снова госпитализировали. При неврологическом обследовании выявлен спастический нижний парапарез, мышечная сила составляла 3 балла в левой и 4 балла в правой ноге, высокие сухожильные рефлексы в ногах и патологические стопные рефлексы. Также наблюдался повышенный уровень белка в ликворе, нормальный уровень IgG. МРТ головного мозга демонстрировало диффузную и симметричную гиперинтенсивность белого вещества без особых отличий от предыдущей МРТ. Атрофия головного мозга была заметной и характеризовалась увеличенными желудочками, и атрофией мозолистого тела. На МР-ангиографии — норма. Вызванные зрительные потенциалы не получены. Учитывая все данные, диагноз был пересмотрен и заменен на МЛД. Было проведено генетическое тестирование: скрининг показал полную делецию экзона 4, новую мутацию p.P220L и уже известную мутацию p.T393S в гене ARSA.
Данный клинический случай поздней манифестации в виде ухудшения зрения не является типичным для позднего МЛД. Однако начало заболевания стоит отчитывать с появления депрессивной симптоматики у пациентки и развития когнитивных нарушений. Конечно, можно было бы приписывать ухудшение обучаемости именно депрессией, однако значительное снижение по диагностическим шкалам дает ясно понять о наличии определенной патологии, выходящей за рамки депрессии.
Конечно, уцепиться за диагноз рассеянного склероза можно было из-за зрительных нарушений и поражения белого вещества, но гораздо больше факторов говорили против этого. Обнаружив отсутствие улучшения на фоне приема глюкокортикоидов, несоответствие МР-картины (типичнейшая картина лейкодистрофии, не вовлекающей U-волокна в патогенетический процесс, никакого усиления сигнала на фоне введения контраста), отсутствие олигоклональных антител, атипичной клинической картины, был рассмотрен другой диагноз — лейкодистрофия. Генетическое исследование помогло уточнить диагноз.
Нейрональный цероидный липофусциноз (НЦЛ) [21]
НЦЛ, также известные как болезнь Баттена, представляют собой группу моногенных наследственных нейродегенеративных заболеваний, которые проявляются в основном в первые 10 лет жизни.
Все пациенты с НЦЛ, за исключением пациентов с редкой врожденной формой (10 типа), имеют нормальное психомоторное развитие до появления первых симптомов. У большинства пациентов начало заболевания отмечается в детском возрасте, но для некоторых он может достигать 60 лет и старше. Основные проявления представляют собой сочетание по крайней мере двух из следующих симптомов: деменция, эпилепсия, ухудшение моторных функций и потеря зрения.
Для взрослой формы НЦЛ существуют критерии, выведенные Американской ассоциацией неврологов [22]:
Клинические критерии (требуется выполнение всех 4 пунктов):
- возраст начала между 12 и 60 годами;
- нормальное развитие и сохранные когнитивные функции к моменту манифестации;
- наличие хотя бы двух следующих критериев:
— эпилептические приступы или миоклонус;
— прогрессивное когнитивное снижение;
— атаксия;
— пирамидные или экстрапирамидные знаки; - наличие симптоматики в анамнезе в течение хотя бы двух лет.
Гистологические критерии:
Достоверные:
— Гранулярные осмиофильные отложения (GRODs, профили отпечатков пальцев, криволинейные профили или прямолинейные комплексы) более чем в одном типе клеток, включая, помимо прочего, эккринные секреторные и протоковые клетки, эндотелиальные клетки, клетки гладких и поперечно-полосатых мышц, нейроны или их аксоны. Профили отпечатков пальцев в гладких мышцах сосудов и перицитах исключаются, поскольку было показано, что они неспецифически накапливаются с возрастом.
Вероятные:
— одна или несколько характерных морфологий (GROD, профили отпечатков пальцев, криволинейные профили или прямолинейные комплексы) в одном типе клеток
И
— цитоплазматические включения с широкодиапазонной автофлуоресценцией или окрашиваемые Люксолом быстрым синим о в хорошо дифференцированных срезах в одном или нескольких из перечисленных выше типов клеток в степени, значительно превышающей ожидаемую для возраста.
ИЛИ
— характерный цитоплазматический запасной материал, то есть субъединица c митохондриальной аденозинтрифосфат синтазой, сапозиновый белок A или сапозиновый белок D.
Возможные:
— Цитоплазматические включения с широкодиапазонной автофлуоресценцией или окрашиваемые Люксолом быстрым синим в хорошо дифференцированных участках в одном или нескольких из вышеуказанных типов клеток, в степени, значительно превышающей ожидаемую для возраста.
Основываясь на вышеописанных критериях, можно допустить постановку следующего диагноза:
- Определенный взрослый НЦЛ:
— все клинические критерии плюс определенный гистологический. - Вероятный взрослый НЦЛ:
— атипичная клиническая картина (в т. ч., более позднее начало) плюс определенные гистологические критерии;
— все клинические критерии плюс вероятная гистология. - Возможный взрослый НЦЛ:
— атипичная клиническая картина плюс вероятная гистология;
— все клинические критерии плюс возможная гистология.
Против взрослого НЦЛ говорит нижеследующее:
- клинические, гистологические, либо молекулярные данные совершенно не соответствуют НЦЛ и предполагают другой диагноз;
- обзор микрофотографий (изображений электронной микроскопии материала) или исходного материала свидетельствует об отсутствии признаков НЦЛ;
- изображения патологии недоступны (в т. ч. при наличии только отчета).
Диагностика ЛБН [1]
Диагностика данной группы заболеваний основывается на биохимических и молекулярно-генетических исследованиях.
Биохимическая диагностика представляет собой измерение функции лизосомных ферментов лейкоцитов крови и биопсии вовлеченных тканей. Когда уровни ферментов опускаются ниже нормального диапазона, конкретный тип ЛБН определяют путем генетического тестирования для выявления мутаций в гене, кодирующем недостающий фермент. Однако ферментные анализы проводятся in vitro на искусственных субстратах и могут неточно отражать активность ферментов относительно естественных субстратов in vivo.
При секвенировании ДНК в случае, если выявленные мутации уже были описаны у других пациентов, это может облегчить прогнозирование состояния пациента. Однако при отсутствии кровного родства между родителями или если они не относятся к популяции высокого риска, пациенты обычно оказываются гетерозиготами, что может изменить привычное соответствие фенотипа генотипу. Некоторые ЛБН, обусловленные нарушением функции интегральных мембранных белков, большинство НЦЛ и болезней с нарушением лизосомных органелл могут быть диагностированы только путем прямого секвенирования генов. Если ранее сообщалось, что выявленные мутации являются патогенетическими, диагноз ЛБН может быть подтвержден. Однако если обнаруживаются новые варианты с неопределенной значимостью, то результаты являются предварительными и должны быть соотнесены с клиническим фенотипом.
Скрининг новорожденных
До 2023 года в большинстве регионов РФ обязательный скрининг новорожденных позволял выявить 5 наследственных заболеваний (адреногенитальный синдром, галактоземию, врожденный гипотиреоз, муковисцидоз, фенилкетонурию). С 31.12.2022 года начал действовать Приказ №274н «Об утверждении порядка оказания медицинской помощи пациентам с врожденными и (или) наследственными заболеваниями». Согласно этому документу, неонатальный скрининг новорожденных расширен до 36 заболеваний (в т. ч., глутаровая ацидемия типы 1 и 2, тирозинемия тип 1, и др.) [24].
Терапия ЛБН
Таблица 4. Этиотропная терапия некоторых ЛБН [1]
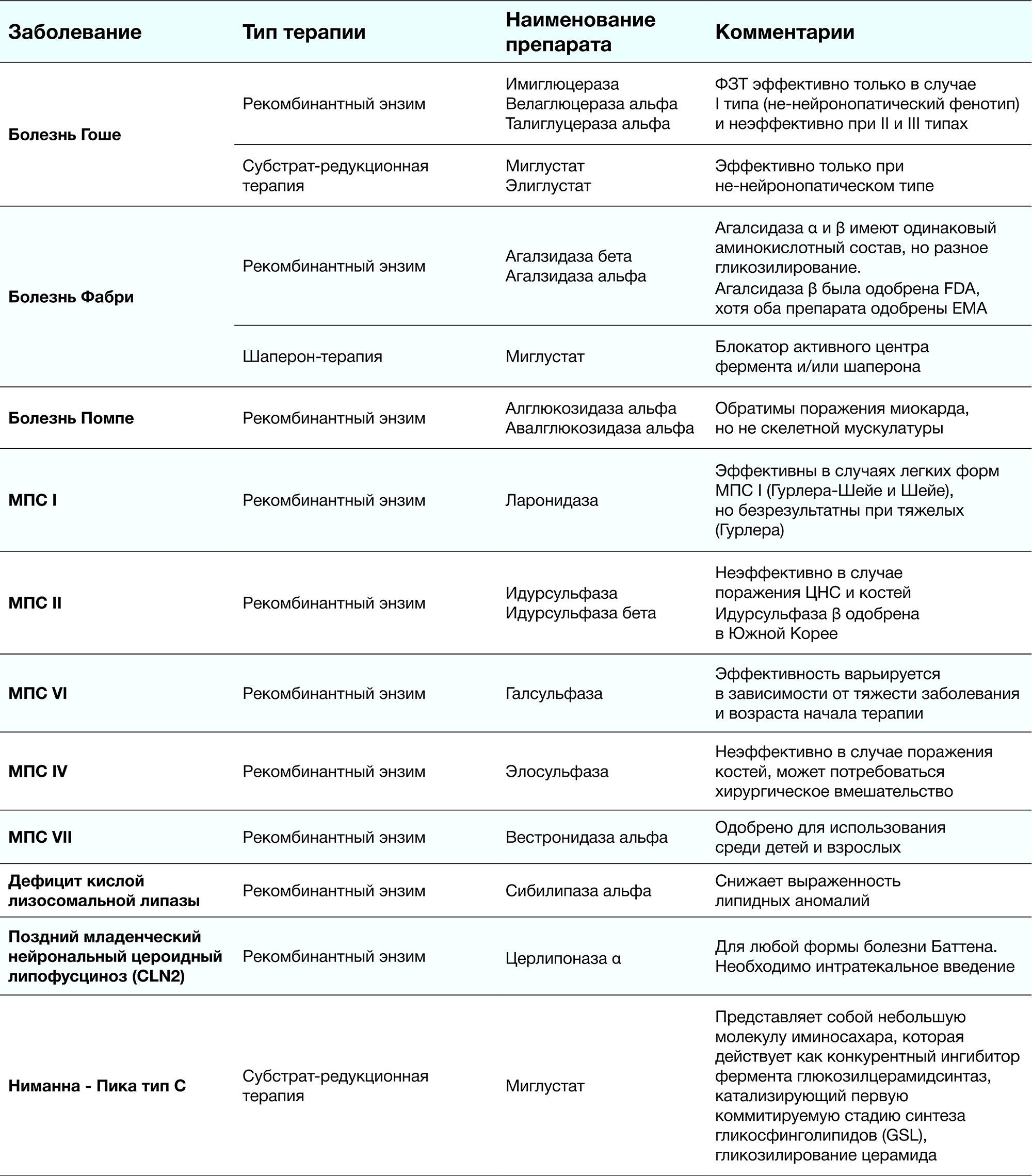
Некоторые ЛБН поддаются лечению: например, дефицит или дефект фермента может быть восполнен экзогенно через ФЗТ, количество накопленного балласта в лизосомах может быть уменьшено с помощью субстрат-редуцирующей терапии (СРТ) либо при шапероновой терапии можно постараться улучшить функцию дефектного фермента. В качестве альтернативы возможны генетические методы лечения (в неопределенном будущем). Хотя в настоящее время для некоторых ЛБН доступно этиотропное лечение (таблица 4), для большинства заболеваний лечение остается симптоматическим. Результаты лечения различаются между ЛБН и сильно варьируют. По мере разработки новых методов лечения может появиться выбор между различными препаратами; если нет медицинских или научных причин для продолжения приема одного лекарства, выбор может основываться на стоимости и/или опыте лечащих врачей. Также имеет место комбинирование лекарственных средств.
С другой стороны, некоторые из представленных выше препаратов не зарегистрированы в РФ и обходятся в десятки миллионов рублей в год на одного пациента, в результате чего вероятность получить терапию крайне мала! Однако через суд можно все-таки добиться закупки данного препарата из регионального бюджета, учитывая жизненные показания [28].
Литература:
- Platt F. M. et al. Lysosomal storage diseases //Nature reviews Disease primers. – 2018. – T. 4. – №. 27. https://doi.org/10.1038/s41572-018-0025-4
- https://dic.academic.ru/dic.nsf/ruwiki/372936
- Oxford Dictionary of Biochemistry and Molecular Biology (2 ed.). Edites by Cammack, R. et al. 2008. https://www.oxfordreference.com/display/10.1093/acref/9780198529170.001.0001/acref-9780198529170-e-11527
- Rajkumar V, Dumpa V. Lysosomal Storage Disease. [Updated 2023 Jul 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024.
- Stirnemann J. et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments //International journal of molecular sciences. – 2017. – Т. 18. – №. 2. – С. 441.
- Alaei MR, Tabrizi A, Jafari N, Mozafari H. Gaucher Disease: New Expanded Classification Emphasizing Neurological Features. //Iran J Child Neurol. – 2019. – T. 13. – №. 1. – C. 7–24.
- https://emedicine.medscape.com/article/951637-clinical#b4
- Regier DS, Tifft CJ, Rothermel CE. GLB1-Related Disorders. // University of Washington, Seattle. – 22 Apr 2021.
- Muthane U. et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases //Movement disorders: official journal of the Movement Disorder Society. – 2004. – Т. 19. – №. 11. – С. 1334-1341.
- Ramani P. K., Sankaran B. P. Tay-sachs disease // StatPearls [Internet]. – StatPearls Publishing, 2023.
- Deik A, Saunders-Pullman R. Atypical presentation of late-onset Tay-Sachs disease. //Muscle Nerve. – 2014. – Т. 49. – №. 5. – С. 768-71.
- Sung A. R., Moretti P., Shaibani A. Case of late-onset Sandhoff disease due to a novel mutation in the HEXB gene //Neurology: Genetics. – 2018. – Т. 4. – №. 4. – С. e260.
- Sheth J. et al. GM2 gangliosidosis AB variant: novel mutation from India–a case report with a review //BMC pediatrics. – 2016. – Т. 16. – С. 1–5.
- Morales A, Anilkumar AC. Glycogen Storage Disease Type II. StatPearls [Internet] 2017.
- Chan J. et al. The emerging phenotype of late-onset Pompe disease: A systematic literature review //Molecular genetics and metabolism. – 2017. – Т. 120. – №. 3. – С. 163-172.
- Peters H. et al. Treatable lysosomal storage diseases in the advent of disease‐specific therapy //Internal Medicine Journal. – 2020. – Т. 50. – С. 5-27.
- Ezgu F. et al. Expert opinion on the recognition, diagnosis and management of children and adults with Fabry disease: a multidisciplinary Turkey perspective //Orphanet journal of rare diseases. – 2022. – Т. 17. – №. 1. – С. 90.
- Sethuraman G, Chouhan K, Kaushal S, Sharma VK. Fabry's disease. //Lancet. – 2011. – Т. 378. – №. 9798. – С. 1254.
- Shaimardanova A. A. et al. Metachromatic leukodystrophy: diagnosis, modeling, and treatment approaches //Frontiers in medicine. – 2020. – Т. 7. – С. 576221.
- Xu L. et al. Case report: Novel arylsulfatase A (ARSA) gene mutations in a patient with adult-onset metachromatic leukodystrophy misdiagnosed as Multiple sclerosis //Frontiers in Neurology. – 2021. – Т. 11. – С. 576881.
- Mole S. E. et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis //The Lancet Neurology. – 2019. – Т. 18. – №. 1. – С. 107-116.
- Berkovic S. F. et al. Diagnosis and misdiagnosis of adult neuronal ceroid lipofuscinosis (Kufs disease) //Neurology. – 2016. – Т. 87. – №. 6. – С. 579-584.
- Приказ Минздрава РФ от 22.03.2006 № 185 «О массовом обследовании новорожденных детей на наследственные заболевания» https://legalacts.ru/doc/prikaz-minzdravsotsrazvitija-rf-ot-22032006-n-185/[1]
- https://roddom.msk.ru/skrining
- https://minzdrav.gov.ru/news/2021/06/01/16743-po-porucheniyu-predsedatelya-pravitelstva-minzdrav-prorabotaet-vopros-o-rasshirenii-neonatalnogo-skrininga-do-36-zabolevaniy[2] https://dalnhospital.3dn.ru/news/neonatalnyj_skrining_novorozhdennykh_v_2024_godu/2024-03-14-645
- https://www.medscape.com/viewarticle/956179
- Wraith J. E., Imrie J. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat //Therapeutics and clinical risk management. – 2009. – С. 877-887.
- https://asi.org.ru/news/2020/03/23/regions-rnd-lekarstva-sud/
