
Ключевые признаки рака

После четверти века стремительного прогресса в области изучения рака было получено большое количество данных, из которых становится понятно, что при раке в геноме происходят динамические изменения. Основой для исследований рака стало открытие мутаций, увеличивающих активность онкогенов и снижающих активность генов, отвечающих за подавление опухолевого процесса. По изменениям в геномах человеческих и животных раковых клеток удалось определить оба класса генов, связанных с раком. (Bishop and Weinberg, 1996).
Кто-то может возразить, что поиск происхождения этой болезни и лекарства от нее будет продолжаться еще более четверти века, по большей части абсолютно таким же образом, что и раньше: и без того слишком затруднительная для понимания научная литература только продолжит усложняться. Но мы ожидаем другого: исследователи этой проблемы будут заниматься наукой совершенно иначе, нежели чем это делалось в течение последних 25 лет. Конечно, основные изменения произойдут на техническом уровне, однако нельзя исключать и фундаментальные изменения научного подхода.
Мы предполагаем, что исследования рака станут более логичными, а описанные в лабораториях и клиниках особенности болезни станут понятны с точки зрения небольшого количества основополагающих принципов. Часть этих принципов систематизируют уже сейчас. В этом эссе мы обсудим закономерности трансформации обычных человеческих клеток в злокачественные. В исследованиях, проведенных за последние несколько десятилетий, выявлено небольшое количество молекулярных, биохимических и клеточных свойств, общих для большей части или, возможно, для всех видов человеческого рака. Возможность такого упрощения напрямую следует из учения о клеточной биологии, которое утверждает, что пролиферация, дифференцировка и гибель всех клеток млекопитающих регулируется с помощью одних и тех же молекулярных механизмов.
Существует множество оснований считать, что онкогенез – многоступенчатый процесс, и каждая ступень отражает генетические изменения, приводящие к постепенному преобразованию обычных человеческих клеток в злокачественные. Частота встречаемости многих видов раковых опухолей зависит от возраста, причем 4 из 7 заболеваний возникают стохастически (Renan, 1993). В ходе патологоанатомического исследования некоторых областей органов были выявлены повреждения, которые, по всей видимости, представляют собой промежуточный этап в процессе трансформации, во время которого клетка проходит через несколько «предзлокачественных» состояний. (Foulds, 1954).
После анализа проведенной работы были сделаны следующие выводы: геном опухолевых клеток страдает как от малых повреждений (точечные мутации), так и от значительных, таких как изменения в хромосомном наборе (e.g., Kinzler and Vogelstein, 1996). Трансформация культивируемых клеток является многоступенчатым процессом: клеткам грызунов требуется как минимум два приобретенных генетических изменения перед тем, как они получат способность к опухолеобразованию, в то время как их человеческие аналоги сложнее подвергнуть трансформации (Hahn et al., 1999). Трансгенная модель онкогенеза неоднократно подтверждала тезис о том, что опухолеобразование у мышей включает в себя множество лимитирующих стадий (Bergers et al., 1998; see Oncogene,1999, R. DePinho and T. E. Jacks, volume 18[38], pp. 5248–5362). Взятые в совокупности сведения о человеческом раке и его моделях на животных служат доказательством того, что процесс развития опухоли формально аналогичен дарвиновской теории эволюции. Наследование генетических изменений, каждое из которых дает то или иное преимущество, приводит к постепенному преобразованию обычных человеческих клеток в раковые (Foulds, 1954; Nowell, 1976).
Список свойств
В нормальных клетках функционируют механизмы, препятствующие развитию рака, в то время как в опухолевых клетках имеются дефекты в регуляторной цепи, контролирующей нормальную пролиферацию клетки и ее гомеостаз. Описано более ста различных типов рака, при этом в определенных органах можно выделить даже подтипы опухолей. Такая сложная структура этого заболевания вызывает много вопросов. Сколько различных регуляторных цепей должно быть разрушено внутри каждого типа клетки-мишени, прежде чем клетка станет раковой? Одинаковые ли регуляторные цепи повреждаются в клетках разных новообразований, возникающих в теле человека? Какие из этих цепей работают автономно, а какие связаны с сигналами, которые клетки получают от тканевого микроокружения? Может ли большая и разнообразная группа ассоциированных с раком генов быть связана с работой небольшой группы регуляторных цепей?
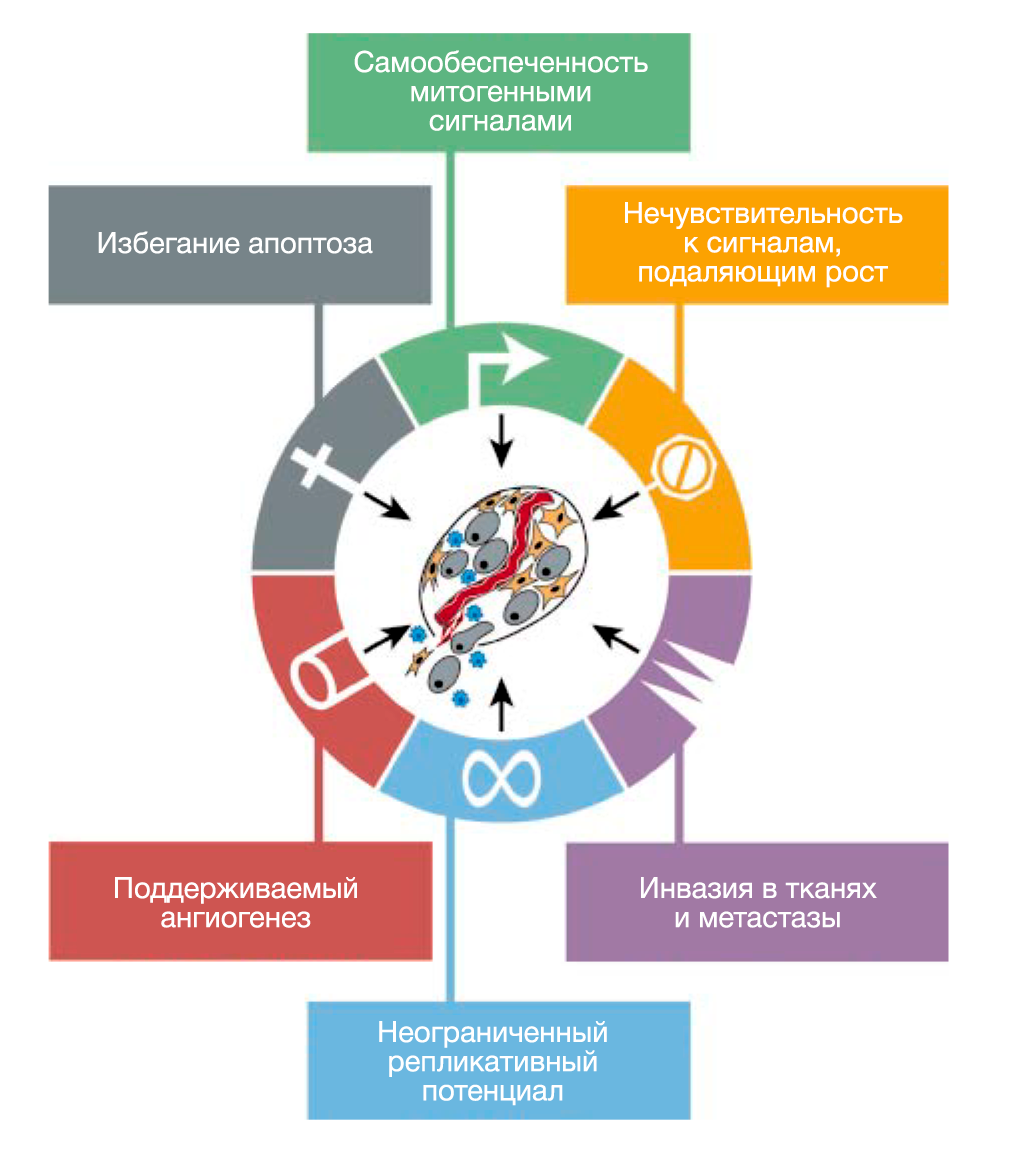
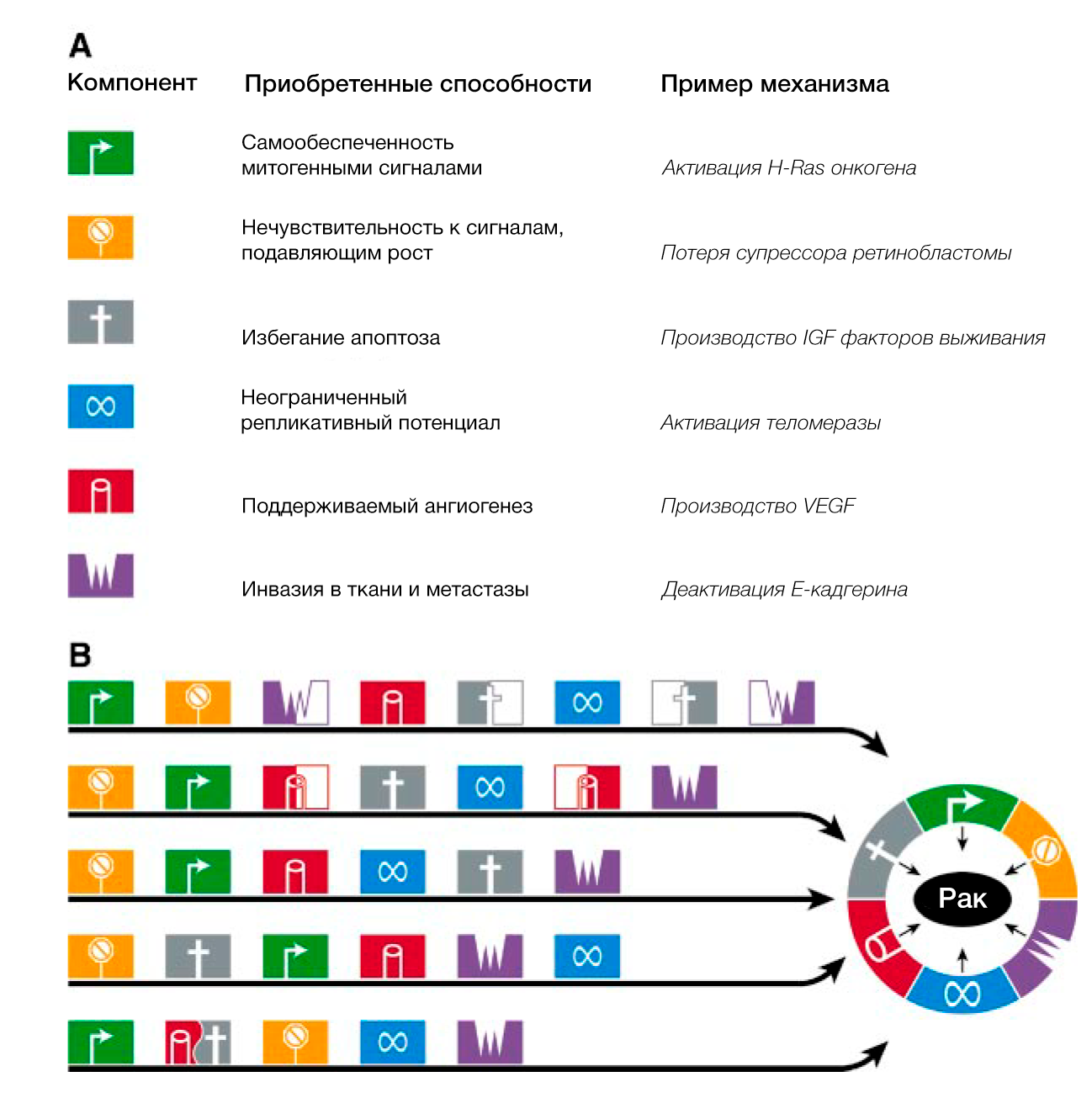
Мы предполагаем, что обширный перечень генотипов раковых клеток — это проявление шести важных изменений в клеточной физиологии, которые в совокупности влияют на рост злокачественности (РИС.1 Приобретенные способности рака). Мы предполагаем также, что большинство или, возможно, все виды рака в ходе своего развития приобретают одни и те же свойства: самообеспеченность сигналами роста, нечувствительность к сигналам ингибиторов роста, уклонение от запрограммированной клеточной смерти (апоптоза), неограниченный репликативный потенциал, поддерживаемый ангиогенез, способность к инвазии в ткани и метастазированию. Каждое из этих физиологических изменений дает новые возможности и свидетельствует об успешном нарушении противоопухолевого защитного механизма, действующего в клетках и тканях. Такое многообразие защитных механизмов может объяснить, почему рак возникает относительно редко на протяжении средней человеческой жизни.
Мы опишем каждое свойство по очереди, иллюстрируя несколькими примерами его функциональное значение и указывая способы, с помощью которых это свойство было приобретено.
Приобретенные способности: самообеспеченность митогенными сигналами
Для того, чтобы обычная клетка перешла из состояния покоя в пролиферативное состояние, ей необходимы митогенные сигналы роста (СР). Сигналы передаются в клетку через трансмембранные рецепторы, которые связывают молекулы разных классов: растворимые факторы роста, компоненты внеклеточного матрикса, молекулы адгезии и взаимодействия с остальными клетками. Насколько нам известно, ни один тип обычных клеток не может пролиферировать без стимулирующего сигнала. Множество раковых онкогенов тем или иным способом имитируют нормальный митогенный сигнал.
Очевидно, что нормальным клеткам митогенные сигналы жизненно необходимы: при культивировании они размножаются только в присутствии подходящего растворимого фактора роста и подходящего субстрата для их интегринов. Совсем по-другому ведут себя раковые клетки, которые неизменно демонстрируют значительно сниженную зависимость от экзогенной стимуляции роста. Опухолевые клетки генерируют множество собственных сигналов роста, тем самым уменьшая свою зависимость от стимулов, исходящих от нормального микроокружения. В результате этого освобождения раковых клеток от необходимости получать экзогенные сигналы нарушается очень важный гомеостатический механизм, который в норме обеспечивает правильное поведение различных типов клеток в ткани.
По причине высокой встречаемости онкогенов, модулирующих независимость от факторов роста, это свойство раковых клеток было описано исследователями первым. На молекулярном уровне можно выделить три основных способа обеспечения такой независимости: изменение внеклеточных сигналов роста, трансмембранных проводников этих сигналов или внутриклеточных путей, которые приводят эти сигналы в действие. Большая часть растворимых факторов роста (РФР) производится одним типом клеток и стимулирует пролиферацию других (гетеротипическая сигнализация), однако многие раковые клетки способны синтезировать РФР, к которым они чувствительны, создавая замкнутый цикл положительной обратной связи (аутокринная стимуляция) (Fedi et al., 1997). Очевидно, что продукция факторов роста раковыми клетками позволяет им не зависеть от факторов роста, продуцируемых другими клетками ткани. В качестве примеров можно привести производство PDGF (Тромбоцитарного Фактора Роста) и TGFα (Трансформирующего Фактора Роста α) клетками глиобластомы и саркомы соответственно (Fedi et al., 1997).
Во время развития опухоли нарушается регуляция поверхностных клеточных рецепторов, осуществляющих трансдукцию митогенных сигналов внутрь клетки. При многих видах рака происходит гиперэкспрессия обладающих тирозинкиназной активностью рецепторов к факторам роста. При избыточной экспрессии рецепторов чувствительность раковых клеток к ФР увеличивается, и они начинают реагировать на фоновую концентрацию ФР, которая в норме не приводит к началу пролиферации (Fedi et al., 1997). К примеру, в клетках раковых опухолей желудка, мозга и молочных желез активируется рецептор к эпидермальному ФР (EGF-R/erbB), тогда как в клетках карцином желудка и молочной железы наблюдается оверэкспрессия рецептора HER2/neu (Slamon et al., 1987; Yarden and Ullrich, 1988). Кроме того, значительная оверэкспрессия рецепторов к ФР может привести к лиганд-независимой передаче сигналов (DiFiore et al., 1987). Лиганд-независимая передача сигналов может также возникать при структурных изменениях рецепторов: например, укороченный вариант рецептора к EGF, лишенный большей части цитоплазматического домена, постоянно активен.
Раковые клетки также могут сменить тип экспрессируемых ими рецепторов внеклеточного матрикса (интегринов), выбирая те, которые передают сигналы роста (Lukashev and Werb, 1998; Giancotti and Ruoslahti, 1999). Эти бифункциональные гетеродимерные рецепторы на поверхности клетки физически связывают клетки с внеклеточными суперструктурами, известными как внеклеточный матрикс (ВКМ). В случае успешного прикрепления интегринов к компонентам ВКМ начинается трансдукция сигнала, что влияет на поведение клетки: в обычной ткани клетка выходит из состояния покоя, а в раковой увеличивается ее подвижность, клетка становится резистентной к апоптозу и входит в активный клеточный цикл. Напротив, если интегринам не удается установить связь с матриксом, клетка утрачивает подвижность, останавливает клеточный цикл и может войти в состояние апоптоза (Giancotti and Ruoslahti, 1999). Как лигандзависимые рецепторы ФР, так и проростовые интегрины после контакта с матриксом могут активировать молекулярный путь SOS-Ras-Raf-MAPK. (Aplin et al., 1998; Giancotti and Ruoslahti,1999).
Наиболее сложны механизмы возникновения изменений во внутриклеточных нисходящих каскадах, проводящих сигналы от активированных лигандзависимых рецепторов к ФР и интегринов. Каскад SOS-Ras-Raf-MAPK играет главную роль в этом процессе. Примерно в 25% человеческих опухолей структура белков Ras нарушена, что позволяет им проводить внутрь клеток большое количество митогенных сигналов, избегая при этом тормозящего воздействия восходящих регуляторов (Medema and Bos, 1993).
Мы подозреваем, что митогенные сигнальные пути нарушены во всех человеческих опухолях. Хотя эту идею пока сложно доказать строго, косвенных догадок имеется предостаточно (Hunter, 1997). Например, в случае карциномы толстой кишки человека — наиболее изученной из всех опухолей — около половины образований несут мутантные онкогены ras (Kinzler and Vogelstein, 1996). Мы предполагаем, что остальные опухоли в кишечнике содержат дефекты в других компонентах сигнальных путей роста, фенокопирующих активацию онкогена ras. Природа этих альтернативных механизмов стимулирования роста остается неясной.
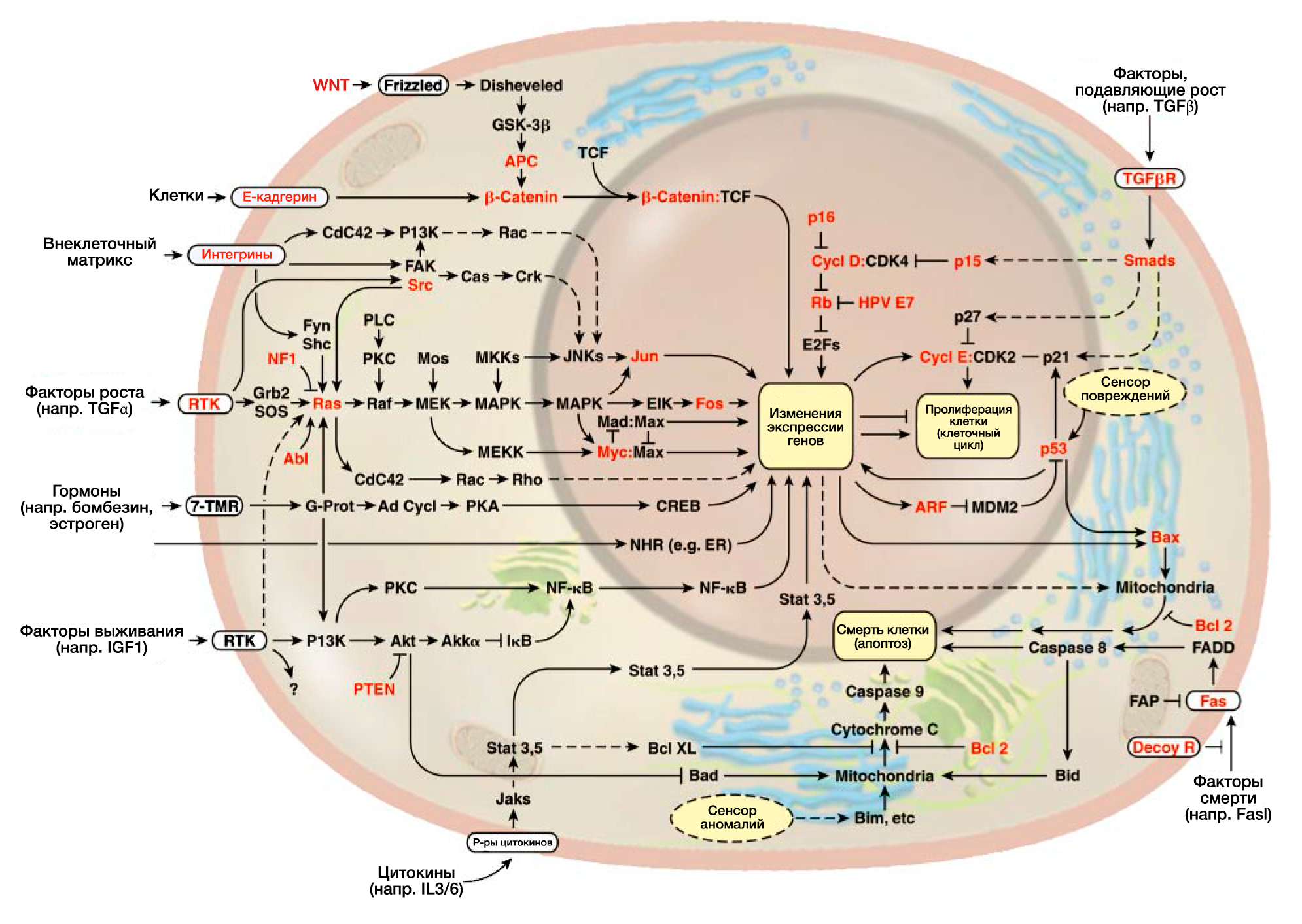
Два десятилетия интенсивного изучения рака позволили составить схему митогенных сигнальных путей в клетках млекопитающих(РИС.2). Регулярно обнаруживаются новые нижестоящие эффекторные пути, которые зависят от центрального митогенного каскада SOS-Ras-Raf-MAPK (Hunter, 1997; Rommel and Hafen, 1998). Этот каскад также связан с другими молекулярными путями с помощью разнообразных перекрестных соединений, и эти перекрестные связи позволяют экзогенным сигналам вызывать в клетке сразу несколько биологических эффектов. Например, прямое взаимодействие белка Ras со способствующей выживанию киназой PL3 позволяет сигналам роста вызвать также сигнал выживания внутри клетки (Downward, 1998).
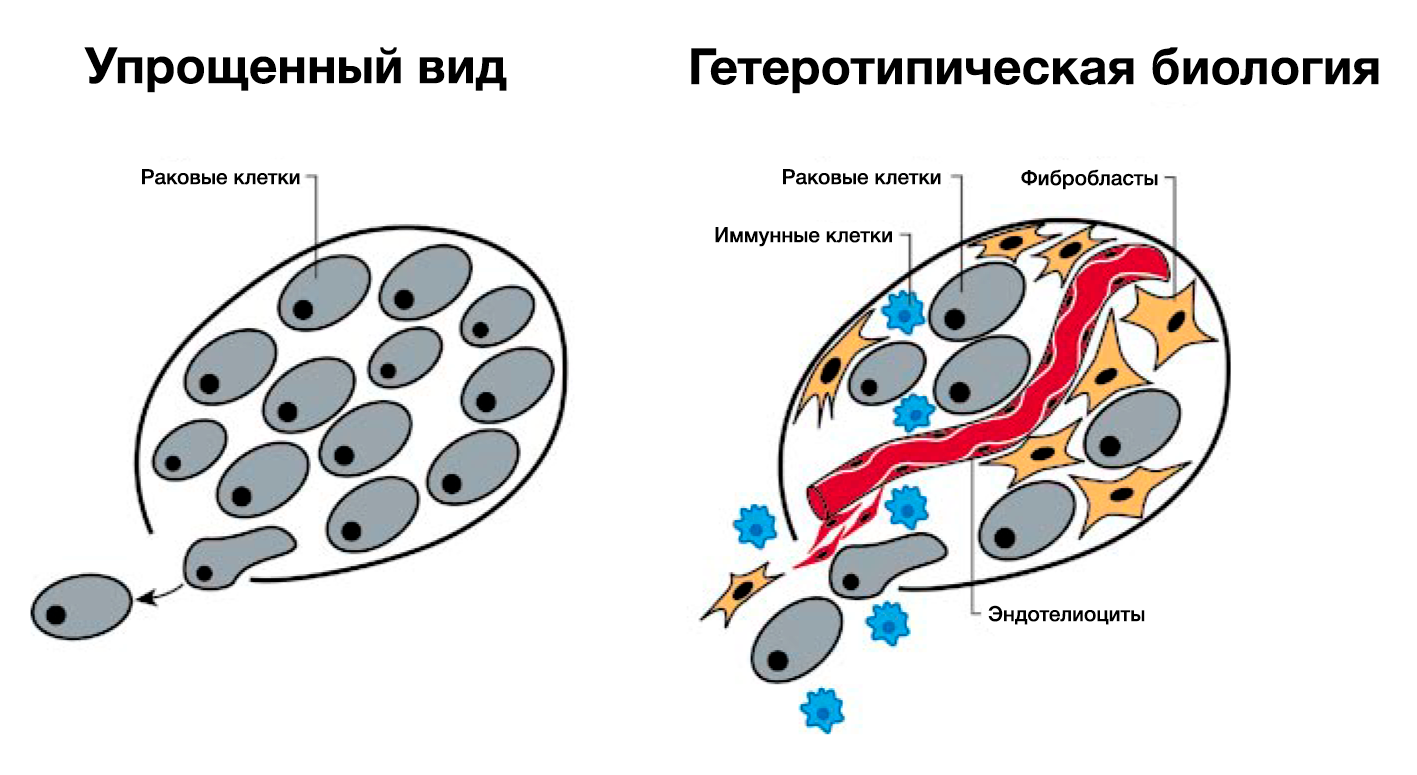
Несмотря на то, что концепция приобретенной автономии РФ вполне удовлетворительна, она чересчур упрощена. Мы традиционно исследовали рост опухоли, обращая экспериментальное внимание на генетически измененные раковые клетки (РИС.3, левая часть). Однако всё более очевидно то, что дерегуляция роста внутри опухоли может быть объяснена только тогда, когда мы поймем, каков вклад вспомогательных клеток, представленных в опухоли обычными клетками, такими как фибробласты и эндотелиоциты, которые играют ключевую роль в процессе пролиферации опухолевой клетки (РИС.3, правая часть). В обычной ткани рост клетки в основном регулируется её соседями (паракринные сигналы) или через системные (эндокринные) сигналы. Межклеточные сигналы роста имеются в подавляющем большинстве человеческих опухолей: фактически все опухоли состоят из разных типов клеток, которые взаимодействуют с помощью гетеротипической передачи сигналов.
Гетеротипическая передача сигналов между разнообразными типами клеток внутри опухоли может в итоге оказаться настолько важной в понимании пролиферации опухолевых клеток, насколько важны автономные механизмы раковых клеток, перечисленные выше. Например, мы подозреваем, что множество сигналов роста происходят из стромальных клеточных компонентов опухоли. Такое мышление изменяет логику приобретенной автономии сигналов роста: успешные опухолевые клетки – клетки, получившие возможность использовать своих обычных соседей путем стимуляции их к высвобождению избыточного количества сигналов роста (Skobe and Fusenig, 1998). Разумеется, в некоторых опухолях эти сотрудничающие клетки могут иметь отклонения от нормы, развиваясь вместе со своими злокачественными соседями и обеспечивая рост последних (Kinzler and Vogelstein, 1998; Olumi et al., 1999). Кроме того, воспалительные клетки, мигрирующие к органам и тканям с неоплазией, могут содействовать раковым клеткам, а не ликвидировать их (Cordon-Cardo and Prives, 1999; Coussens et al., 1999; Hudson et al., 1999). Это еще один пример взаимодействия нормальных клеток с раковыми, в результате которого злокачественные клетки получают такую необходимую способность, как независимость от митогенных сигналов.
Приобретенные способности: невосприимчивость к сигналам, подавляющим рост
В обычной ткани множественные антипролиферативные сигналы поддерживают клеточный покой и тканевой гомеостаз; эти сигналы включают в себя растворимые ингибиторы роста и иммобилизованные ингибиторы, встроенные во внеклеточный матрикс и поверхность окружающих клеток. Эти сигналы, действующие с помощью тех же механизмов, что и митогенные, попадают во внутриклеточную сигнальную цепь через трансмембранные рецепторы на поверхности клетки.
Сигналы, подавляющие рост, могут препятствовать пролиферации двумя различными механизмами. Во-первых, клетки могут быть выведены из активного пролиферативного цикла и переведены в фазу покоя (G0), из которой они в будущем смогут выйти только тогда, когда изменение внеклеточного сигнала позволит им это сделать. Во-вторых, клетки могут быть введены в постмитотическое состояние, в котором они приобретут определенные свойства, связанные с терминальной дифференцировкой, и навсегда потеряют свой пролиферативный потенциал.
Зарождающиеся раковые клетки должны обходить эти антипролиферативные сигналы. Большая часть механизма (в особенности компоненты, регулирующие выход клетки из фазы цикла G1), который позволяет обычным клеткам реагировать на антипролиферативные сигналы, связана с длительностью клеточного цикла. Клетки следят за окружающей их микросредой на протяжении всего периода роста и на основе полученных сигналов решают пролиферировать, оставаться в покое или переходить в постмитотическое состояние. На молекулярном уровне все или почти все антипролиферативные сигналы направляются через белок ретинобластомы (pRb) и два родственных белка p107 и p130. В гипофосфорилированном состоянии pRb препятствует пролиферации путём блокирования функции транскрипционных факторов E2F, которые контролируют экспрессию групп генов, необходимых для перехода от фазы G1 к фазе S (Weinberg, 1995).
Нарушение pRb-пути освобождает E2F и, таким образом, позволяет клетке пролиферировать. Это приводит к размножению клеток, нечувствительных к подавляющим рост факторам, которые обычно блокируют выход из фазы G1 посредством молекулярного пути pRb. Наиболее полно описаны эффекты растворимой сигнальной молекулы TGFβ, но мы предполагаем, что другие антипролиферативные факторы также будут передавать сигнал по этому пути. Различными способами, большинство из которых все еще неизвестно, TGFβ предотвращает фосфорилирование и деактивацию pRb; таким образом, TGFβ блокирует переход из фазы G1 к последующим фазам клеточного цикла. В некоторых типах клеток TGFβ подавляет экспрессию гена c-myc, который регулирует фазу цикла G1, однако механизмы этого действия еще не изучены (Moses et al.,1990). TGFβ напрямую контролирует синтез белков p15INK4B и p21, которые блокируют комплекс циклин:CDК, отвечающий за фосфорилирование pRb (Hannon and Beach, 1994; Datto et al., 1997).
В разных типах человеческих опухолей цепь передачи сигналов через pRb, регулируемая TGFβ и другими внешними факторами, может быть нарушена различными способами (Fynan and Reiss, 1993). Некоторые клетки перестают отвечать на молекулу TGFβ в ходе подавления экспрессии их TGFβ-рецепторов, другие же синтезируют мутантные нефункциональные рецепторы (Fynan and Reiss, 1993; Markowitz et al., 1995). Цитоплазматический белок Smad4, который проводит сигналы из лиганд-активируемых TGFβ рецепторов к последующим белкам в цепи, может быть элиминирован путём мутации кодирующего его гена (Schutte et al., 1996). Может быть удален локус, кодирующий белок p15INK4B(Chin et al., 1998). Кроме того, непосредственная мишень p15INK4B CDK4 может перестать реагировать на его ингибирующее действие из-за мутаций, в результате которых одни аминокислотные остатки замещаются другими в INK4A/B-взаимодействующем домене; в результате комплексы циклин D:CDK4 свободно инактивируют pRb путем гиперфосфорилирования (Zuo et al., 1996). В результате функциональный белок pRb, являющийся конечной мишенью в этом молекулярном пути, может быть потерян в ходе мутации гена. С другой стороны, в некоторых опухолях, индуцированных ДНК-вирусами, например, в карциноме шейки матки, функция pRb снижена в результате действия вирусных онкобелков, таких как онкобелок Е7 вируса папилломы человека (Dyson et al., 1989). Кроме того, раковые клетки могут прекращать экспрессию интегринов и других клеточных молекул адгезии, которые передают сигналы, подавляющие рост, в пользу тех, которые передают стимулирующие рост сигналы. Возможно, подавляющие рост сигналы, действующие при адгезии, также влияют на путь pRb. В результате подавляющие рост пути сходятся на Rb, а цикл клеточного деления тем или иным образом нарушается в большинстве человеческих опухолей, что обуславливает потерю механизмов онкосупрессии при раке.
Клеточная пролиферация зависит не только от уклонения от цитостатических сигналов, подавляющих рост. Наши ткани также сдерживают деление клеток, заставляя клетки необратимо перейти в постмитотическое, дифференцированное состояние, используя для этого разнообразные механизмы, которые еще не до конца изучены. Очевидно, что опухолевые клетки используют различные способы, чтобы избежать терминальной дифференцировки. Один из способов избежать дифференцировки – использовать непосредственно онкоген c-myс, который кодирует транскрипционный фактор. В ходе нормального развития Myc стимулирует рост вместе с другим фактором – Max, конкурируя с альтернативными комплексами Max с группой транскрипционных факторов Mad; комплекс Mad-Max производит сигналы, активирующие дифференцировку (Foley and Eisenman, 1999). Однако оверэкспрессия онкобелка c-Myc, которая наблюдается во многих видах опухолей, может обратить этот процесс, сдвигая баланс в пользу комплексов Myc-Max, тем самым тормозя дифференцировку и содействуя росту. В ходе канцерогенеза в толстой кишке человека дезактивация молекулярного пути APC/b-катенин блокирует переход энтероцитов в крипте ободочной кишки в дифференцированное, постмитотическое состояние (Kinzler and Vogelstein, 1996). Аналогично, во время сборки вируса птичьего эритробластоза онкоген erbA предотвращает терминальную дифференцировку эритроцитов (Kahn et al., 1986).
Взаимосвязь антиростовых и индуцирующих дифференцировку сигналов с основными механизмами регулировки клеточного цикла все еще плохо изучена. Однако существование схемы сигнальной трансдукции, подавляющей рост, очевидно (РИС.2), так как уход от нее необходим для развития рака.
Приобретенные способности: избежать апоптоза
Способность популяций опухолевых клеток активно размножаться определяется не только скоростью клеточной пролиферации, но также и скоростью клеточной убыли. Запрограммированная смерть клетки – апоптоз – представляет собой основную причину этой убыли. В ходе исследований на мышиных моделях и культурах клеток, а так же описательных анализов стадий карциногенеза по биопсии человеческих опухолей были получены свидетельства того, что приобретенная сопротивляемость апоптозу является признаком всех или почти всех типов рака.
Накопленные за последнее десятилетие знания указывают на то, что программа апоптоза присутствует почти во всех типах клеток по всему организму в скрытой форме. После запуска с помощью разных физиологических сигналов эта программа разворачивается в точно продуманный ряд шагов. В промежутке 30 – 120 минут: разрушаются клеточные мембраны, цитоплазматический и ядерный скелеты, экструдируется цитозоль, деградируют хромосомы и фрагментируются ядра. В конце деградировавшие остатки клетки исчезают после поглощения окружающими клетками ткани, обычно в пределах 24 часов (Wyllie et al., 1980).
Компоненты системы, обеспечивающей апоптоз, можно грубо разделить на две группы: эффекторы и сенсоры. Сенсоры ответственны за мониторинг условий внеклеточной и внутриклеточной среды. Нормальное либо ненормальное состояние условий среды влияет на то, будет ли клетка жить или же умрет. Эти сигналы регулируют второй класс компонентов, которые функционируют в качестве эффекторов апоптоза. Сенсоры включают себя рецепторы на поверхности клетки, которые связывают факторы выживания и смерти. В качестве примера пар лиганд/рецептор, передающих сигнал выживания, можно назвать IGF-1/IGF-2 с их рецептором — IGF-1R а также IL-3 и соответствующий рецептор IL-3R (Lotem and Sachs, 1996; Butt et al., 1999). Сигналы смерти проводятся через лиганд FAS, который связывается с FAS рецепторами, и TNFα, который связывается с TNF-R1 (Ashkenazi and Dixit, 1999). Внутриклеточные сенсоры контролируют клеточное благополучие и активируют молекулярный путь смерти, если обнаружат аномалии, включающие в себя повреждения ДНК, дисбаланс в передаче сигналов, вызванный действием онкогенов, недостаточность факторов выживания или гипоксию (Evan and Littlewood, 1998). Кроме того, жизнь большинства клеток частично поддерживается адгезией клетки к клеточному матриксу и к остальным клеткам, что служит сигналом к выживанию, потеря которого индуцирует апоптоз (Ishizaki et al., 1995; Giancotti and Ruoslahti, 1999). Как растворимые, так и иммобилизованные регуляторные сигналы, вероятно, отражают необходимость ткани поддерживать составляющие её клетки в подходящей архитектурной конфигурации.
Множество сигналов, вызывающих апоптоз, сходится на митохондриях, которые отвечают на проапоптотические сигналы освобождением цитохрома С, мощного катализатора апоптоза (Green and Reed, 1998). Члены семейства белков Bcl-2, которые имеют и проапоптотическую (Bax, Bak, Bid, Bim), и антиапоптотическую (Bcl-2, Bcl-XL, Bcl-W) функцию, действуют сообща, управляя митохондриальным сигналингом смерти, высвобождая цитохром С. Белок-онкосупрессор p53 может вызвать апоптоз, повышая экспрессию проапоптотического Bax в ответ на повреждение ДНК; Bax в свою очередь стимулирует высвобождение цитохрома С митохондриями.
Окончательные эффекторы апоптоза включают в себя набор внутриклеточных протеаз, названных каспазами (Thornberry and Lazebnik, 1998). Две инициирующие каспазы – 8 и 9 — активируются рецепторами смерти, такими как FAS или цитохром С, высвобожденными митохондрией. Эти начальные каспазы вызывают активацию дюжины или большего количества эффекторных каспаз, которые выполняют программу смерти путем избирательного разрушения субклеточных структур, органелл и генома.
Идея того, что апоптоз препятствует развитию рака, была впервые предложена в 1972 г., когда Kerr, Wyllie and Currie описали стремительно развивающийся массивный апоптоз в клеточных популяциях гормон-зависимых опухолей, возникающий после отмены гормонов (Kerr et al., 1972). Открытие апрегуляции онкогена bcl-2 через хромосомную транслокацию в фолликулярной лимфоме (обзор Korsmeyer, 1992) и выявление того, что этот онкоген проявляет антиапоптотическую активность (Vaux et al., 1988), положили начало изучению апоптоза в раковых клетках на молекулярном уровне. Ген bcl-2, коэкспрессированный с онкогеном myc в трансгенной мыши, мог содействовать формированию В-клеточной лимфомы путем повышения выживаемости лимфоцитов, а не последующей стимуляцией myc—индуцированной пролиферации (Strasser et al., 1990); более того, 50% редких лимфом, возникающих в bcl-2 трансгенных мышах, обладают соматическими транслокациями активирующих c-myc, повышая регуляцию Bcl-2 и c-Myc во время онкогенеза (McDonnell and Korsmeyer, 1991).
Однако понимание взаимодействия myc-bcl-2 появилось позже, во время изучения эффектов онкогена myc в фибробластах, культивированных в среде с низким содержанием сыворотки. Недостаток сыворотки индуцировал массивный апоптоз в myc-экспрессирующих клетках; апоптоз можно было нейтрализовать экзогенными факторами выживания (e.g., IGF-1), принудительной гиперэкспрессией Bcl-2 или родственного ему белка Bcl-XL или путем нарушения передачи сигнала смерти в пути FAS (Hueber et al., 1997). Взятая в совокупности информация указывает на то, что программа клеточного апоптоза может быть вызвана гиперэкспрессированным онкогеном. Действительно, устранение клеток, несущих активированные онкогены, путем апоптоза является основным средством, благодаря которому мутантные клетки постоянно удаляются из тканей организма.
Другие примеры подтверждают мнение, что апоптоз – основное препятствие для рака, которое необходимо обойти. У трансгенных мышей, у которых опухолевый супрессор pRb был функционально деактивирован в сосудистом сплетении, возникали медленно растущие микроскопические опухоли с высоким уровнем апоптоза; дополнительная деактивация белка p53, опухолевого супрессора, компонента апоптотической сигнальной цепи, приводила к развитию стремительно растущих опухолей, содержащих небольшое количество апоптических клеток (Symonds et al., 1994). Роль внеклеточных факторов выживания может быть проиллюстрирована на примере прогрессирования заболевания у трансгенных мышей, склонных к опухолям панкреатических островков. Если экспрессированный ген IGF-2, активируемый в этом молекулярном пути, нокаутируется у трансгенной мыши, рост и прогрессирование опухоли сокращаются, о чем свидетельствует появление сравнительно маленьких доброкачественных опухолей с высоким уровнем апоптоза (Christofori et al., 1994). Отсутствие IGF-2 в этих клетках не влияло на скорость пролиферации, что позволяет четко определить это вещество как фактор выживания. Взятые в совокупности данные подтверждают, что нарушение работы различных компонентов апоптического механизма может существенно повлиять на динамику роста опухоли, что объясняет, почему этот механизм инактивирован во время развития опухоли.
Резистентность раковых клеток к апоптозу может быть приобретена различными способами. Конечно, наиболее часто встречаются потери проапоптотического регулятора в ходе мутации в гене-онкосупрессоре p53. В результате функциональная деактивация продукта, белка p53, наблюдается более чем в 50% видов человеческого рака и приводит к удалению ключевого компонента — сенсора повреждения ДНК, который может вызывать апоптотический эффекторный каскад (Harris, 1996). Сигналы, вызванные другими аномалиями, включая гипоксию и гиперэкспрессию онкогенов, также частично направляются в апоптотический каскад через p53; когда p53 не выполняет свою функцию, их работа также нарушена (Levine, 1997). Кроме того, в значительной доле человеческих опухолей, вероятно, предотвращение апоптоза обусловлено активацией молекулярного пути PI3 киназа–AKT/PKB, который передает антиапоптотический сигнал выживания. Эта цепь передачи сигналов выживания может быть активирована внеклеточными факторами, такими как IGF-1/2 или IL-3 (Evan and Littlewood, 1998), внутриклеточными сигналами, исходящими от Ras (Downward, 1998), или нехваткой опухолевого супрессора pTEN, фосфатазы фосфолипидов, которая обычно подавляет сигнал выживания от протеинкиназы В (Cantley and Neel, 1999). В последнее время был выявлен еще один механизм подавления сигнала смерти FAS в большей части клеточных линий карцином легких и карцином толстой кишки: апрегуляция несигнального рецептора-ловушки для FAS лиганда, связывающего этот лиганд и предотвращающего его взаимодействие с рецептором (Pitti et al., 1998). Мы предполагаем, что фактически все раковые клетки содержат изменения, которые позволяют избегать апоптоза.
Теперь возможно изложить предварительную схему трансдукции сигналов апоптоза (РИС.2); хотя схема неполная, очевидно, что большей части регуляторных и эффекторных компонентов хватает с избытком. Эта избыточность имеет большое значение для развития новейших видов антиопухолевой терапии: если опухолевые клетки теряют одни проапоптотические компоненты, они, вероятно, могут сохранить их аналоги. Мы предвидим, что новые технологии помогут обнаружить апоптотические молекулярные пути, все еще действующие в определенных видах раковых клеток, и что новые лекарства активируют взаимодействие между еще неповрежденными компонентами параллельных апоптотических молекулярных путей в опухолевых клетках, что приведет к восстановлению апоптотического защитного механизма со значительным терапевтическим эффектом.
Приобретенные способности: неограниченный репликативный потенциал
Три приобретенные способности (самообеспеченность митогенными сигналами, нечувствительность к сигналам, подавляющим рост, и резистентность к апоптозу) приводят к разобщению программы роста клетки и сигналов окружающей среды. В принципе, этих нарушений в программе пролиферации должно быть достаточно, чтобы обеспечить условия для появления клеточных популяций, которые составляют макроскопические опухоли. Однако исследования последних тридцати лет указывают на то, что это приобретенное нарушение взаимодействия клеток само по себе не обеспечивает экспансивный рост опухоли. Все или почти все виды клеток млекопитающих несут внутреннюю, автономную программу, лимитирующую пролиферацию. Эта программа, по-видимому, действует независимо от передачи сигналов от клетки к клетке через молекулярные пути, описанные выше. Ее уничтожение также необходимо для того, чтобы клоны клеток увеличили опухоль, угрожающую жизни, до макроскопического размера.
Ранние работы Hayflick демонстрируют, что клетки в культуре обладают конечным репликативным потенциалом (обзор Hayflick, 1997). После того, как клеточные популяции проходят через определенное число делений, они перестают расти – этот процесс называется старением. Старения культивированных человеческих фибробластов можно избежать путем выведения из строя их белков pRb и p53 -опухолевых супрессоров, позволяющих клеткам продолжить размножение, пока они не войдут в следующее состояние, называемое кризисом. Кризисное состояние характеризуется массивной клеточной смертью, кариотипическими нарушениями, связанными со слиянием хромосом конец-в-конец и случайным появлением варианта (1 из 107), когда клетка получает способность размножаться без ограничений. Это приобретение называется иммортализация (Wright et al., 1989).
Большая часть опухолевых клеток, растущих в культуре, оказывается иммортализованной. Таким образом, способность бесконечно реплицироваться — фенотипическое свойство, которое необходимо клеткам для злокачественного роста и которое они приобретают in vivo во время развития опухоли (Hayflick, 1997). Как следствие, в какой-то момент во время многоступенчатого процесса развития опухоли предраковые клеточные популяции исчерпывают свой лимит удвоений и могут стать полностью раковыми только преодолев барьер смертности и приобретя возможность бесконечно реплицироваться.
Исследования культивируемых клеток указывают на то, что нормальные клетки человеческого организма обладают лимитом в 60-70 удвоений. По правде говоря, эти числа не имеют особого смысла, если рассматривать гибель клеток как препятствие для формирования рака: 60-70 делений позволят клонам опухолевых клеток увеличить свое количество настолько, что они смогут превысить количество обычных клеток в человеческом теле. При обобщении оценок уровней пролиферации и апоптоза в определенных человеческих опухолях (Wyllie et al., 1980) и в моделях трансгенных мышей (Symonds et al., 1994; Shibata et al., 1996; Bergers et al., 1998) парадокс разрешается: развивающиеся предраковые и раковые популяции клеток страдают от массивного апоптоза, а следовательно, имеет место быть определенная убыль клеток, сопутствующая их накоплению. Таким образом, количество клеток в опухоли не позволяет оценить количество делений, необходимых для образования этого числа клеток, а лимит делений нормальной клетки является одним из барьеров на пути развития раковой опухоли.
«Счетчик» клеточных поколений был открыт в последнем десятилетии: концы хромосом — теломеры, которые состоят из нескольких тысяч повторений коротких последовательностей, состоящих из 6 пар оснований. Потеря 50-100 пар оснований теломерной ДНК на концах каждой хромосомы во время клеточного цикла расценивается как одно поколение. Это постепенное сокращение происходит в результате неспособности ДНК-полимеразы полностью реплицировать 3’-концы хромосомной ДНК во время каждой S фазы. Постепенная потеря длины теломер в ходе последующих циклов репликации в конечном итоге становится причиной потери их способности к защите концов хромосомной ДНК. При наличии незащищенных теломерами концов хромосомы соединяются конец-в-конец, что приводит к нарушению кариотипа, ассоциируемому с кризисом, и практически неизбежной гибели клетки (Counter et al., 1992).
Очевидно, практически во всех видах злокачественных клеток происходит поддержание длины теломер на должном уровне (Shay and Bacchetti, 1997); 85% — 90% таких клеток преуспевают в этом благодаря повышению экспрессии фермента теломеразы, который добавляет гексануклеотидные повторы на концы хромосом (Bryan and Cech, 1999), тогда как оставшиеся клетки нашли способ активации механизма, называемого АПУТ (альтернативный путь удлинения теломер), при котором длина теломер поддерживается на должном уровне с помощью межхромосомной рекомбинации (Bryan et al., 1995). Восстановление хромосом посредством того или иного механизма делает возможным безграничное деление клеток. В большинстве нормальных человеческих клеток оба этих механизма подавляются, благодаря чему такие клетки не обладают неограниченным репликативным потенциалом.
Роль теломеразы в иммортализированных клетках может быть продемонстрирована непосредственно путем эктопической экспрессии фермента в клетках, что может привести к передаче неограниченного репликативного потенциала еще не сенесцентным клеткам ранних пассажей in vitro (Bodnar et al., 1998; Vaziri and Benchimol, 1998). Экспрессия этого фермента в клетках поздних пассажей, практически готовых ко вступлению в кризисное состояние, позволяет им продолжать пролиферацию без каких-либо признаков кризиса (Counter et al., 1998; Halvorsen et al., 1999; Zhu et al., 1999). В ходе исследований мышей, страдающих от нехватки функции теломеразы, были получены дополнительные сведения о важности поддержания длины теломер для развития рака. Например, мыши с нокаутированным вариантом ингибитора клеточного цикла p16INK4A в гомозиготном состоянии склонны к развитию опухолей, особенно когда вступают в контакт с канцерогенами; возникающие опухоли имеют сравнительно высокую теломеразную активность. Когда p16INK4A-null мыши, страдающие от нехватки теломеразы, контактировали с канцерогенами, частота опухолей у них была снижена одновременно с существенным укорочением теломер и появлением кариотипических нарушений (Greenberg et al., 1999).
В то время как совершенно очевидно, что поддержание длины теломер является ключевым компонентом способности к безграничной репликации, остается невыясненным вопрос по поводу другого компонента – избегания клеточного старения. Феномен старения был первоначально замечен как задержка пролиферации клеток в растущей культуре и ассоциирован с подсчетом клеточных делений (Hayflick, 1997). В последнее время было обнаружено, что состояние старения индуцируется в определённых культивированных клетках в ответ на высокий уровень экспрессии генов, таких как активированный онкоген ras (Serrano et al., 1997).
Приведенные выше наблюдения могут противоречить тому, что старение так же, как и апоптоз, отражает защитный механизм, который может быть активирован путем укорочения теломер или с помощью конфликтующих сигналов, вызывающих рост, которые заставляют клетки с отклонениями от нормы необратимо перейти в G0-подобное состояние и тем самым делают их неспособными к дальнейшей пролиферации. Если это так, избегание старения in vivo представляет собой важный шаг в развитии опухоли, который необходим для последующего приближения к нарушению кризисного барьера. Но мы считаем, что возможен и другой вариант: старение может быть артефактом, проявляющимся при искусственном культивировании клеток, который не отражает фенотип клетки внутри живой ткани и не представляет собой препятствие для развития опухоли in vivo. Разрешение этого вопроса будет важно для полного понимания приобретенного безграничного репликационного потенциала.
Приобретенная способность: устойчивый ангиогенез
Кислород и питательные вещества, поставляемые сосудистой системой, необходимы для функционирования клетки и поддержания жизни, и именно поэтому практически все клетки ткани вынуждены располагаться в пределах 100 мкм от капилляра. Во время органогенеза эта близость обеспечивается скоординированным ростом сосудов и паренхимы. После того, как ткань сформируется, рост новых кровеносных сосудов – ангиогенез – является преходящим и тщательно регулируется. Из-за этой зависимости от окружающих капилляров, вполне вероятно, пролиферирующие клетки внутри ткани должны иметь внутреннюю способность увеличивать рост кровеносных сосудов. Но эксперимент говорит об обратном. Клетки внутри аномальных пролиферативных поражений изначально страдают от нехватки ангиогенных способностей, которая сокращает их возможности к экспансии. Чтобы прогрессировать в размере, зарождающиеся новообразования должны развивать свои ангиогенные способности (Bouck et al., 1996; Hanahan and Folkman, 1996; Folkman, 1997).
Ангиогенез контролируется путем уравновешивания положительных и отрицательных сигналов. Один класс этих сигналов транспортируется растворимыми факторами и их рецепторами, размещенными на поверхности эндотелиальных клеток; не менее важную роль играют интегрины и адгезивные молекулы, служащие посредниками в связях клетка-матрикс и клетка-клетка. Примером сигналов, инициирующих ангиогенез, служит фактор роста эндотелия сосудов (VEGF), а также кислый и основной факторы роста фибробластов (FGF1/2). Каждый из них связывается с трансмембранными тирозинкиназными рецепторами, экспрессируемыми эндотелиальными клетками (Fedi et al., 1997; Veikkola and Alitalo, 1999). Типичный ингибитор ангиогенеза – тромбоспондин-1, который связывается с CD36, трансмембранным рецептором эндотелиальных клеток, спаренным с внутриклеточной Src-подобной тирозинкиназой (Bull et al., 1994). Сегодня известно более двух дюжин ангиогенных факторов и такое же число белков-ингибиторов.
Передача сигналов интегринами также способствует поддержанию этого баланса. Покоящиеся клетки сосудов экспрессируют один класс интегринов, в то время как клетки растущих капилляров экспрессируют другой. Конкуренция сигналов из второго класса интегринов может препятствовать ангиогенезу (Varner and Cheresh, 1996; Giancotti and Ruoslahti, 1999), что подчеркивает значительный вклад клеточной адгезии в ангиогенез (Hynes and Wagner, 1996). Внеклеточные протеазы структурно и функционально связаны с проангиогенными интегринами и регулируют проникающую способность ангиогенных эндотелиальных клеток (Stetler-Stevenson, 1999).
Экспериментальные доказательства важности возникновения и поддержания ангиогенеза в опухолях обширны и убедительны (Bouck et al., 1996; Hanahan and Folkman, 1996; Folkman, 1997). История началась почти 30 лет назад, когда Folkman и его коллеги продемонстрировали необходимость ангиогенеза для экспансивного роста опухолевых эксплантатов в биологических исследованиях in vivo (обзор Folkman, 1997). Молекулярные доказательства принципа появились, когда выяснилось, что антитела против VEGF способны нарушать образование новых сосудов и замедлять рост подкожных опухолей у мышей (Kim et al., 1993), как и блокирующая проведение сигнала форма VEGF receptor 2 (flk-1) (Millauer et al., 1994); эти данные подтолкнули к исследованию специфичных ингибиторов VEGF/VEGF-R, которые теперь находятся на последних стадиях клинических испытаний.
Важная роль ангиогенеза подтверждается способностью большого количества антиангиогенных веществ нарушать рост опухолевых клеток, введенных мышам подкожно (Folkman, 1997). Опухоли, возникающие у предрасположенных к раку трансгенных мышей, также чувствительны к ангиогенным ингибиторам (Bergers et al., 1999).
Умение индуцировать и поддерживать ангиогенез, видимо, приобретается на отдельной ступени (или нескольких ступенях) развития опухоли через «ангиогенный сдвиг» покоящихся клеток сосудов. Когда многоступенчатый туморогенез был проанализирован на трех моделях трансгенных мышей, в каждом случае было обнаружено, что ангиогенез активировался во время промежуточных повреждений, прежде чем разовьется зрелая опухоль. Аналогично можно выявить ангиогенез в предзлокачественных поражениях шейки матки, молочной железы и кожи (меланоцитов) (Hanahan and Folkman, 1996); мы полагаем, что ангиогенез можно определить на ранних стадиях во многих видах человеческого рака. Эти сведения, взятые в совокупности со сведениями об эффектах ангиогенных ингибиторов, указывают на то, что неоваскуляризация – это предвестник быстрой клеточной экспансии, приводящей к появлению макроскопических опухолей.
Опухоли, вероятно, активируют «ангиогенный сдвиг” путем изменения равновесия между индукторами ангиогенеза и их антагонистами — ингибиторами (Hanahan and Folkman, 1996). Нарушенная транскрипция генов – распространенная стратегия для сдвига равновесия. Множество данных об опухолях свидетельствуют о повышенной экспрессии VEGF и/или FGF по сравнению с их обычными тканевыми аналогами. По другим данным, может подавляться экспрессия эндогенных ингибиторов, таких как тромбоспондин-1 или β-интерферон. Тем не менее, вероятны оба варианта и, разумеется, они взаимосвязаны в некоторых опухолях (Singh et al., 1995; Volpert et al., 1997).
Механизм, лежащий в основе изменения баланса между ангиогенными регуляторами, остаётся не совсем понятным. В одном задокументированном примере было обнаружено, что белок-онкосупрессор p53 положительно регулирует ингибитор тромбоспондин-1 в некоторых типах клеток. Вследствие этого потеря функций p53, которая имеет место быть в большей части человеческих опухолей, может стать причиной падения уровня тромбоспондина-1, освобождая эндотелиальные клетки от его ингибиторного эффекта (Dameron et al., 1994). Экспрессия гена VEGF контролируется сложной системой. Например, активация онкогена ras или потеря гена-онкосупрессора VHL в определенных типах клеток становится причиной апрегуляции экспрессии VEGF (Rak et al., 1995; Maxwell et al., 1999).
На регуляцию ангиогенеза могут влиять и протеазы, которые могут контролировать биодоступность ангиогенных активаторов и ингибиторов. Таким образом, разнообразные протеазы могут высвобождать bFGF, который хранится в ECM (Whitelock et al., 1996), в то время как плазмин, проангиогенный компонент системы свертывания, может отщепить от себя ингибитор ангиогенеза, называемый ангиостатин (Gately et al., 1997). Координированная экспрессия про- и антиангиогенных сигнальных молекул и их модуляция протеолизом отражает сложную гомеостатическую регуляцию обычного тканевого ангиогенеза и сосудистой целостности.
Очевидно, что опухолевый ангиогенез является привлекательной терапевтической мишенью, общей для всех или почти всех типов опухолей. В следующем десятилетии будет открыт целый набор регуляторных ангиогенных молекул, экспрессируемых разными видами опухолей и их предшественниками. Использование более сложных моделей мышей позволит определить роль каждого из этих регуляторов и рассмотреть молекулярный механизм, управляющий их производством и активностью. Доступные к настоящему моменту сведения указывают на то, что разные типы опухолевых клеток используют разные молекулярные стратегии, чтобы активировать ангиогенный сдвиг. Это поднимает следующий вопрос: будет ли достаточно единственной антиангиогенной молекулы для лечения всех видов опухолей или необходимо несколько таких, каждая из которых будет отвечать за разные программы ангиогенеза, используемые различными классами человеческих опухолей?
Приобретенные способности: инвазия в ткани и метастазы
Рано или поздно в ходе развития большинства видов человеческого рака первичная опухолевая масса порождает клетки, которые отделяются от нее, вторгаются в соседние ткани и перемещаются оттуда в более отдаленные места организма, где могут успешно основывать новые колонии. Эти отдаленные поселения опухолевых клеток – метастазы – причина 90% смертей от рака (Sporn, 1996). Способность к инвазии и метастазированию позволяет раковым клеткам покинуть основную опухолевую массу и колонизировать новые места в теле, где, по крайней мере первоначально, нутриенты и пространство не ограничены. Новые сформировавшиеся метастазы возникают как совокупность раковых клеток и обычных поддерживающих клеток из питающей ткани. Так же как и формирование основной опухолевой массы, успех инвазии и метастазирования зависит от всех остальных пяти приобретенных отличительных способностей. Но какие дополнительные клеточные изменения облегчают приобретение этих последних способностей в ходе туморогенеза?
Инвазия и метастазирование — чрезвычайно сложные процессы, и их генетические и биохимические детерминанты остаются не совсем понятными. На механическом уровне это два тесно связанных процесса, они являются проявлениями одной общей способности раковых клеток и тесно связаны друг с другом. Оба используют одинаковые стратегии, в том числе изменения структурной связи клеток с их микроокружением и активацию внеклеточных протеаз.
В клетках, способных к инвазии и метастазированию, нарушается функция нескольких классов белков, участвующих в прикреплении клеток к тканевому окружению. К этим белкам относятся и молекулы межклеточной адгезии (ММА) – в частности, члены семейства иммуноглобулинов и Ca2+-зависимых кадгеринов, которые служат посредниками в межклеточном взаимодействии, и интегрины, которые связывают клетки и субстраты внеклеточного матрикса. Все эти «адгезивные» взаимодействия передают в клетку регуляторные сигналы (Aplin et al., 1998). Наиболее изучены нарушения клеточной адгезии, в которых основную роль играет Е-кадгерин — гомотипическая молекула межклеточного взаимодействия, повсеместно экспрессируемая эпителиальными клетками. Соединение смежных клеток Е-кадгерином приводит к передаче подавляющих рост и прочих сигналов через цитоплазматические контакты с β-катенином во внутриклеточные сигнальные пути, в том числе реализующие свое действие через транскрипционный фактор Lef/Tcf (Christofori and Semb, 1999). Е-кадгерин, по-видимому, не выполняет свою функцию в большинстве эпителиальных опухолей в результате мутационной деактивации генов Е-кадгерина или β-катенина, репрессии транскрипции или протеолиза внеклеточного домена кадгерина (Christofori and Semb, 1999). Принудительная экспрессия Е-кадгерина в культивированных раковых клетках и в моделях канцерогенеза в трансгенных мышах ухудшает способность к инвазии и метастазированию, в то время как интерференция с функцией Е-кадгерина усиливает обе способности (Christofori and Semb, 1999). Таким образом, Е-кадгерин служит широко действующим супрессором инвазии и раковых метастазов, и его функциональное устранение представляет ключевой шаг в получении новой способности.
Изменение экспрессии CAM суперсемейства иммуноглобулинов также играет важную роль в процессе инвазии и метастазирования (Johnson, 1991). Самый яркий пример включает в себя N-CAM: экспрессия этого белка переключается с высокоадгезивной изоформы на низкоадгезивную или даже отталкивающую изоформу в опухолях Вильмса, нейробластомах и мелкоклеточном раке легких (Johnson, 1991; Kaiser et al., 1996), при этом при инвазивных раках поджелудочной железы и колоректальном раке экспрессия в целом понижается (Fogar et al., 1997). Эксперименты на трансгенных мышах подтверждают функциональную роль обычных адгезивных форм N-CAM в подавлении метастазирования (Perl et al., 1999).
Очевидно, что в инвазивных и метастатических клетках меняется экспрессия интегринов. При вторжении и метастазировании раковые клетки сталкиваются с различными типами микроокружения, где они могут взаимодействовать с новыми для себя компонентами межклеточного матрикса. Таким образом, успешная колонизация новых мест (как близлежащих, так и отдалённых) требует адаптации, которая достигается путем изменения спектра α и β субъединиц интегрина в мигрирующих клетках. Эти перестановки приводят к экспрессии различных подтипов интегринов (которых больше 22), имеющих различный аффинитет к определенным субстратам. Таким образом, клетки карциномы облегчают инвазию путём переключения экспрессии интегринов с тех, которые специфичны ВКМ в нормальном эпителии, на другие классы (например, α3β1 и αVβ3), которые предпочтительно связывают деградировавшие стромальные компоненты, произведенные внеклеточными протеазами (Varner and Cheresh, 1996; Lukashev and Werb, 1998). Принудительная экспрессия субъединиц интегрина в культивированных клетках может препятствовать инвазивным и метастатическим реакциям или индуцировать их, причем эти рецепторы играют центральную роль в данных процессах (Varner and Cheresh, 1996).
Попытки объяснить клеточные биологические эффекты интегринов с точки зрения небольшого числа механистических правил провалились из-за большого количества различных генов интегринов и еще большего количества гетеродимерных рецепторов, полученных при комбинаторной экспрессии разных α и β рецепторных субъединиц, а также большого количества свидетельств сложности сигналов, получаемых от цитоплазматических доменов этих рецепторов (Aplin et al., 1998; Giancotti andRuoslahti, 1999). Всё еще имеются сомнения, что эти рецепторы играют центральную роль в возможности инвазии в ткани при метастазировании.
Второй основной параметр, определяющий способность к инвазии и метастазированию, являет собой внеклеточные протеазы (Coussens and Werb, 1996; Chambers and Matrisian, 1997). Когда гены протеазы активируются, а гены ингибиторов протеазы подавляются, неактивные зимогенные формы протеазы превращаются в активные ферменты. Протеазы, разрушающие матрикс, связаны с клеточной поверхностью, так как синтезируются вместе с трансмембранным доменом, связываясь с особыми рецепторами протеаз или ассоциируясь с интегринами (Werb, 1997; Stetler-Stevenson, 1999). Можно предположить, что связь активных протеаз с клеточной поверхностью может способствовать инвазии раковых клеток в окружающую строму по стенкам кровеносных сосудов и через обычные эпителиальные клеточные слои. Несмотря на это, трудно приписать функции отдельных протеаз исключительно этой способности, учитывая их роль в ангиогенезе (Stetler Stevenson, 1999) и передаче сигналов роста (Werb, 1997; Bergers and Coussens, 2000), которые, в свою очередь, прямо или косвенно вносят вклад в инвазивные/метастатические способности.
Сложность заключается в том, что множество типов клеток экспрессируют протеазы. Во многих видах карцином протеазы, разрушающие матрикс, продуцируются не эпителиальными раковыми клетками, а скорее стромальными и клетками воспаления (Werb, 1997); однажды произведенные этими клетками, они могут находиться под контролем клеток карциномы. Например, определенные раковые клетки вызывают экспрессию урокиназы (uPA) в сокультивированных стромальных клетках, которая затем привязывается к рецептору урокиназы (uPAR), экспрессированному раковыми клетками (Johnsen et al., 1998).
Активация внеклеточных протеаз, изменение аффинитета кадгеринов, CAM и интегринов — явно необходимые условия для получения способности к инвазии и метастазированию. Однако регуляторные цепи и молекулярные механизмы, которые определяют эти переходы, остаются невыясненными и на данный момент кажутся различными в разных тканевых средах. Приобретенная способность к инвазии и метастазированию представляет собой последний рубеж в исследовании рака. Мы считаем, что развитие аналитических методов позволит вскоре построить комплексные профили экспрессии функционально активных протеаз, интегринов и CAMs в разнообразных типах рака как перед, так и после приобретения инвазивных и метастатических возможностей. Задача будет заключаться в применении новых сведений об инвазивности и метастазировании клеток для развития эффективных терапевтических стратегий.
Новые особенности: нестабильность генома
Приобретение шести вышеперечисленных способностей в ходе развития опухоли создаёт дилемму. Имеющиеся данные свидетельствуют о том, что большинство из них приобретается прямо или косвенно в ходе изменений в геноме раковых клеток. Но мутация конкретных генов — неэффективный процесс из-за непрекращающегося поддержания целостности генома сложным комплексом репарирующих ДНК ферментов. Эти “команды по обслуживанию генома” стремятся к тому, чтобы последовательность информации ДНК оставалась нетронутой. Кариотипический порядок гарантируется еще одним механизмом: так называемые чекпоинты срабатывают в критические моменты жизни клетки, особенно во время митоза. В совокупности эти системы обеспечивают низкую частоту мутаций, настолько редкую, что множественные мутации, присутствующие в геноме опухолевой клетки, весьма маловероятны на протяжении жизни человека.
Тем не менее, в человеческой популяции рак появляется очень часто, вызывая споры, связанные с тем, что геном опухолевых клеток должен приобрести повышенную изменчивость для того, чтобы продолжить рост опухоли до достижения кульминации хотя бы за несколько десятилетий (Loeb, 1991). Чтобы объяснить эту возросшую изменчивость, исследователи нарушали работу определенных компонентов «систем-смотрителей» (Lengauer et al., 1998). Наиболее выделяющаяся часть этих систем – белок-онкосупрессор p53, который вызывает остановку клеточного цикла в ответ на нарушение ДНК, чтобы позволить ей восстановиться, или вызывает апоптоз, если ущерб невосполним. Очевидно, что функция p53 в сигнальных путях нарушения ДНК теряется в большинстве, если не во всех видах рака (Levine, 1997). Более того, растущее число других генов, участвующих в считывании и восстановлении нарушенной ДНК или обеспечении правильной хромосомной сегрегации в ходе митоза, похоже, теряются в различных типах рака, что указывает на онкосупрессорную функцию этих генов. Потеря их функций приводит к геномной нестабильности и вариабельности и появлению мутантных клеток с определенными преимуществами. Интересно, что последние свидетельства говорят, что апоптоз также играет роль в геномной нестабильности, так как ДНК в составе апоптотических телец может быть инкорпорирована в близлежащие клетки при фагоцитозе (Holmgren et al., 1999), внося генетическое разнообразие в любой тип клеток в опухоли. Мы выделяем эту приобретенную особенность отдельно от других шести приобретенных способностей, ассоциированных с раковыми фенотипом и физиологией: она является средством, которое позволяет развивающимся популяциям предзлокачественных клеток приобрести эти шесть способностей.
Альтернативные пути малигнизации
Пути, по которым идут клетки прежде чем стать злокачественными, весьма разнообразны. Внутри одного класса опухолей мутации определенных генов-мишеней, таких как ras или p53, могут наблюдаться только в определенном наборе опухолей, гистологически идентичных остальным. Кроме того, мутации в определенных онкогенах или генах-онкосупрессорах могут возникать рано в одних опухолях и поздно в других. Как следствие, приобретение биологических способностей, таких как устойчивость к апоптозу, поддерживаемый ангиогенез и бесконечный репликативный потенциал, осуществляется в разное время. Соответственно, последовательность приобретения этих способностей сильно различается в зависимости от типа опухоли (Рис. 4). Более того, в одних опухолях одни и те же генетические события приводят к обретению единственной способности, в то время как в других – целого набора. Тем не менее, мы уверены, что независимо от порядка шагов на пути к озлокачествлению, в конце концов клетки приобретают ключевые способности, общие для всех или почти всех типов рака.
Синтез
Большая часть информации, которую мы имеем на данный момент, получена в ходе исследований искусственно выращенных в культурах раковых клеток и их молекулярных компонентов. Упрощая патогенез рака и представляя его как автономный процесс внутри раковых клеток, мы закрываем глаза на реальный процесс опухолеобразования in vivo: развитие рака зависит от гетеротипических взаимодействий зарождающихся раковых клеток и их нормальных соседей. Более того, практически все зрелые опухоли и их метастазы представляют собой смесь клеток, активно взаимодействующих, чтобы поддерживать опухолевый рост (Рис. 3). Эта новая концепция биологии рака серьезно повлияла на экспериментальное изучение этого заболевания. Последующее изучение патогенеза рака будет все больше зависеть от исследований гетеротипичных клеточных культур in vitro и улучшенных мышиных моделей in vivo. В будущем такие системы позволят нам создать понятные карты сетей сигналов роста в опухоли. Трудность заключается в открытии всех сигналов, которыми обмениваются клетки различных типов, сосуществующие в симбиозе в опухолевой массе, и их эффектов на каждый тип клеток.
Наша способность анализировать индивидуальные опухоли на генетическом и биохимическом уровнях также претерпит серьезные изменения. Описание недавно диагностированных опухолей с точки зрения генетических дефектов, лежащих в основе туморогенеза, возможно лишь в перспективе. Тем не менее, мы считаем, что через 10 или 20 лет диагностика соматических повреждений, присутствующих в геноме раковых клеток, станет рутинной процедурой. К тому времени получение полногеномных профилей экспрессии опухолевых клеток также станет обычным делом. Только получив всю эту информацию, удастся доказать наше предположение, что все типы клеток опухолей человека приобретают шесть способностей, описанных здесь.
Мы предполагаем, что удастся значительно лучше понять роль наследуемых аллелей в предрасположенности к раку и в патогенезе этого заболевания. На настоящий момент понимание клеточного взаимодействия унаследованных генов-модификаторов онкогенов и генов-онкосупрессоров с соматически поврежденными генами остается недостаточным; гены-модификаторы могут, в принципе, работать в любом типе клеток внутри опухоли или вообще в организме, тогда как классические раковые гены, в основном, действуют в самих раковых клетках. Эти пробелы будут заполнены отчасти благодаря новым технологиям в информатике, которые позволят нам интерпретировать огромное количество генетической информации, которая вскоре будет получена при автоматическом секвенировании. Новые технологии помогут нам рационализировать сложные взаимодействия аллелей по части систематики формирования опухоли по типу, предложенному здесь.
Концепции функций раковых клеток также сильно изменятся. Через десять лет мы сможем точно предсказать поведение раковой клетки так, как мы предсказываем поведение интегрированной электронной цепи по работе ее составляющих частей – соединяющих компонентов, каждый из которых ответственен за получение, обработку и передачу сигнала согласно детерминированному набору правил. Через два десятилетия мы сможем создать полную «интегрированную цепь клетки» поверх ее текущего наброска. Затем мы сможем применить инструменты математического моделирования, чтобы объяснить, как генетические повреждения перепрограммируют цепь в каждом типе клеток, что приведет к манифестации рака.
Когда мы будем полностью понимать механизм развития рака, прогноз и лечение станут рациональной наукой, которая сейчас не признается терапевтами. Станет возможно с точностью выяснить, почему определенные схемы лечения и противораковые средства успешны или не успешны. Мы предвидим, что будут созданы противораковые препараты, воздействующие на различные ключевые способности раковых клеток; в совокупности со сложными диагностическими технологиями комбинация препаратов позволит побеждать как зарождающиеся, так и зрелые опухоли — недостижимая сегодня цель. Однажды, нам кажется, биология и лечение рака – в настоящий момент смесь биологии, генетики, гистопатологии, биохимии, иммунологии и фармакологии – станут наукой со строгой и логичной структурой, какая наблюдается в химии или физике.
