
Влияние глюкокортикоидов на иммунную систему

Введение
Эндогенные глюкокортикоиды играют важную роль в различных физиологических процессах, включая обмен веществ, развитие и воспаление. Синтетические глюкокортикоиды используются для лечения многих заболеваний иммунной системы c 1948 года. Активно изучаются механизмы, лежащие в основе иммунодепрессивных свойств этих гормонов. Широко известно, что глюкокортикоиды оказывают плейотропное действие на иммунную систему. Тем не менее детальная картина действия глюкокортикоидов на клеточном и молекулярном уровнях остаётся выясненной не до конца. В этом обзоре рассмотрены результаты нескольких десятилетий интенсивных (и часто противоречивых) исследований, в которых изучалось взаимодействие между эндокринной реакцией на стресс и иммунной системой.
Глоссарий
- Лимфолитики — вещества, вызывающие гибель лимфоцитов.
- Стероидогенез — ферментативное превращение холестерола в стероидные гормоны.
- Гипоталамо-гипофизарно-надпочечниковая система (ГГН-система) — система из 3 органов, получающая сигналы от эндокринной, нервной и иммунной систем и контролирующая физиологический ответ на стресс.
- Сумоилирование — посттрансляционная модификация с помощью малых убиквитин-подобных модификационных белков (Small Ubiquitin-like Modifier (SUMO) proteins).
- Иммунофилины — члены семейства высококонсервативных цитозольных изомераз, многие из которых имеют неизвестные клеточные функции.
- Рецепторы распознавания паттернов (PRR) — трансмембранные и цитозольные рецепторы, которые распознают поврежденные и/или связанные с патогенами молекулярные структуры.
- Скавенджер рецепторы — представители подкласса рецепторов распознавания паттернов, функцией которых является участие в процессах идентификации и очистки от клеточного мусора и нежелательных соединений.
- Позитивная селекция — процесс, при котором тимоциты, которые экспрессируют Т-клеточные рецепторы, связывающиеся с белками ГКГС (главного комплекса гистосовместимости), остаются сохранными во время развития Т-лимфоцитов.
- Негативная селекция — процесс, при котором тимоциты, экспрессирующие самореактивные Т-клеточные рецепторы, подвергаются апоптозу.
В 1948 году в клинике Мейо (штат Миннесота, США) пациенту с ревматоидным артритом начали ежедневные инъекции «соединения E» — синтетической версии стероидного гормона, выделенного из надпочечников животных. В течение 3 дней пациент не имел практически никаких симптомов. В 1950 году за открытие «надпочечных кортикальных гормонов» Филиппу Хенчу, Эдварду Кендаллу и Тадеусу Райхштейну была вручена Нобелевская премия по физиологии и медицине. Чудодейственному препарату, соединению Е, было уделено большое внимание, оно упоминалось как глюкокортикоид или кортикостероид, но длительное клиническое применение не поощрялось ввиду неблагоприятных побочных эффектов (вставка 1). Тем не менее глюкокортикоиды до сих пор остаются основой для лечения воспалительной и аутоиммунной патологий и используются в качестве иммунодепрессантов после трансплантации органов и в качестве лимфолитиков в химиотерапии.
На протяжении десятилетий клиническое применение глюкокортикоидов опережало наше механистическое понимание их иммунодепрессивных свойств, а исследование регуляции иммунной системы, опосредованной глюкокортикоидами, и сейчас продолжает оставаться областью интенсивного изучения по нескольким причинам. Во-первых, способы глюкокортикоидного воздействия многообразны. Классически связывание глюкокортикоидов с глюкокортикоидными рецепторами активирует или подавляет транскрипцию генов, а глюкокортикоиды, согласно некоторым исследованиям [1], регулируют до 20% генома. Однако, исследования последних лет выявили и другие механизмы, лежащие в основе глюкокортикоидной активности, включая негеномные эффекты лигандных глюкокортикоидных рецепторов, «перекрестные помехи» глюкокортикоидных рецепторов с другими факторами транскрипции и, возможно, даже рецептор-независимые эффекты. Во-вторых, глюкокортикоидные рецепторы экспрессируются почти во всех клетках, но функциональные эффекты глюкокортикоидов различаются в зависимости от типа клеток. Таким образом, физиологические результаты глюкокортикоидных сигналов представляют собой целый калейдоскоп клеточно-специфичных эффектов.
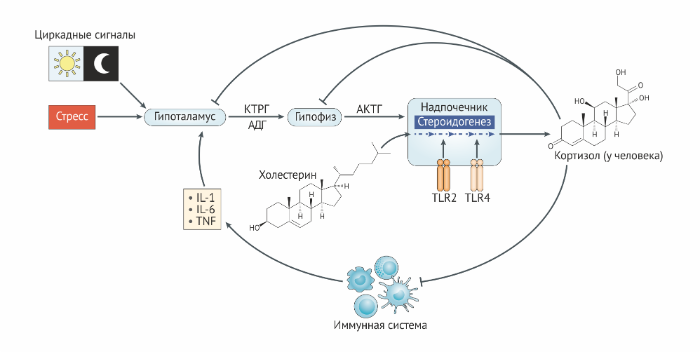
Исследования с истощением популяций клеток и линиями мышей с отсутствием специфических глюкокортикоидных рецепторов позволили изучить специфические эффекты глюкокортикоидов in vivo (вставка 2). Наконец, существуют данные, что глюкокортикоиды способствуют иммунному ответу при определённых условиях, что часто опускается в рамках клинической догмы, согласно которой глюкокортикоиды действуют исключительно как иммунодепрессанты.
Существует огромное количество литературы по теме регуляции иммунной системы глюкокортикоидами. Здесь мы рассматриваем обновлённую информацию о влиянии глюкокортикоидов на канонические иммунные процессы — например, на продукцию медиаторов воспаления, миграцию лейкоцитов, развитие лимфоцитов и передачу сигналов с рецепторов антигенов, — с целью создания пособия для тех, кто интересуется взаимодействием нейроэндокринного стресса и иммунной системы. Мы начнем с краткого обзора синтеза стероидов и глюкокортикоидных рецепторов; следующие разделы будут посвящены обсуждению глюкокортикоид-зависимой регуляции адаптивного иммунитета, врождённого иммунитета и воспаления, а закончим рабочей моделью, которая объясняет как иммунопотенцирующее, так и иммуносупрессивное действие глюкокортикоидов.
Продукция глюкокортикоидов
Стероидогенез
Эндогенные глюкокортикоиды (кортизол у людей и кортикостерон у грызунов) имеют важное значение для жизни, они регулируют мириады физиологических процессов как на этапах развития, так и во взрослом организме. Как и все стероидные гормоны, глюкокортикоиды синтезируются из холестерина в процессе стероидогенеза, происходящего в митохондриях [2]. Синтез глюкокортикоидов происходит в коре надпочечников, хотя местная продукция глюкокортикоидов также была обнаружена в тимусе, кишечнике и коже [3]. Как показано на рис. 1, продукция глюкокортикоидов в надпочечниках запускается при активации гипоталамо-гипофизарно-надпочечниковой системы (ГГН-система), которая представляет собой нервно-эндокринный «эпицентр», координирующий физиологические ответы на внешние раздражители. Глюкокортикоиды, высвобождаемые в кровь, оказывают системное действие путём связывания с глюкокортикоидными рецепторами, присутствующими в клетках по всему организму, включая клетки гипоталамуса и гипофиза, которые являются одним из звеньев отрицательной обратной связи, контролирующей продукцию глюкокортикоидов (рис.1). Примечательно, что первые догадки о влиянии глюкокортикоидов на иммунную систему были получены у пациентов с синдромом Кушинга (который характеризуется избытком глюкокортикоидов, симптомы и патофизиология коротко на Вставке 1) и пациентов с болезнью Аддисона (которая характеризуется дефицитом глюкокортикоидов).
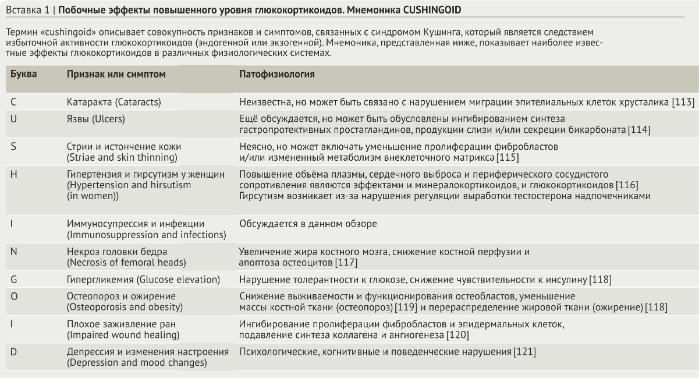
Психологический стресс, физическое напряжение и повреждение тканей активируют ГГН-систему, которая, в свою очередь, запускает быструю и интенсивную продукцию глюкокортикоидов. Гипоталамус также стимулируется интерлейкином 1 (IL1), фактором некроза опухоли (TNF — tumour necrosis factor) и интерлейкином 6 (IL6) [4] (рис.1). Цитокин-индуцированная активация ГГН-системы способствует секреции глюкокортикоидов, которая, как подробно описано ниже, сильно подавляет экспрессию провоспалительных цитокинов. Таким образом, вторая петля обратной связи обеспечивает взаимодействие воспалительного ответа с ГГН-системой (рис. 1). Недавние исследования показали, что прямая активация Toll-like рецепторов 2 и 4 (TLR2 и TLR4) в клетках коры надпочечников индуцирует продукцию глюкокортикоидов, и это указывает на воспалительный путь активации продукции глюкокортикоидов в ГГН-систему (рис.1).
ГГН-система также связывает продукцию глюкокортикоидов с циркадианными и ультрадианными ритмами [6] (рис.1). Хотя амплитуды циркадных и ультрадианных колебаний в стероидогенезе не столь выражены, как те, что связаны с ответом на стресс, эти небольшие изменения в концентрациях глюкокортикоидов по-прежнему модулируют иммунный ответ и миграцию лейкоцитов [7]. Колебания в количестве лимфоцитов крови у людей, например, обратно коррелируют суточными колебаниями секреции глюкокортикоидов, что приводит к сокращению количества циркулирующих Т-клеток на 40% в ночное время как результат изменения их тканевого депонирования.
Местная регуляция активности глюкокортикоидов
Системные изменения концентрации глюкокортикоидов управляются стероидогенезом в надпочечниках, в то время как местная активность глюкокортикоидов регулируется внеклеточными связывающими белками и внутриклеточными ферментами. Кортикостероид-связывающий глобулин (CBG — corticosteroid-binding globulin) инактивирует кортизол, оставляя только около 5% его циркулирующей биоактивной формы. CBG важен для системного распределения глюкокортикоидов через кровообращение, но он также может иметь специфическую тканевую роль в доставке глюкокортикоидов. Например, нейтрофильная эластаза расщепляет CBG, тем самым высвобождая биоактивные глюкокортикоиды в очагах воспаления [9]. Внутри клеток 11β-гидроксистероиддегидрогеназные ферменты (11βHSD1 и 11βHSD2) регулируют взаимопревращение биоактивного кортизола и неактивного кортизона. Воспалительные сигналы, включая TNF и IL1β, модулируют экспрессию ферментов 11βHSD, таким образом изменяя клеточную чувствительность к эндогенным глюкокортикоидам [10].
Глюкокортикоиды в клинической практике
Самым первым глюкокортикоидом, использованным в клинической практике, был синтетический кортизон (соединение Е, описанное выше; отметим, что внутри клеток кортизон превращается в кортизол с помощью 11βHSD1). Хотя его применение было эффективным при лечении ревматоидного артрита, длительная терапия кортизоном вызывает нарушение минерального баланса и задержку жидкости за счёт активации минералокортикоидных рецепторов в почках. Многолетний фармацевтический поиск синтетических глюкокортикоидов, которые имеют минимальное минералокортикоидное действие, привёл к разработке нескольких аналогов, использующихся в клинике сегодня, включая преднизон, беклометазон и флутиказон [11]. Каждый аналог глюкокортикоидов имеет специфические фармакологические свойства, включая жирорастворимость, период полувыведения и минералокортикоидную активность. В целом синтетические глюкокортикоиды являются более сильными иммунорегуляторами, чем кортизол, поскольку они не подвержены эндогенным механизмам ингибирования и инактивации кортизола, таким как связывание с CBG и инактивация 11βHSD. Более того, синтетические глюкокортикоиды обладают более высокой аффинностью к глюкокортикоидным рецепторам и меньшей к минералокортикоидным по сравнению с эндогенными глюкокортикоидами, тем самым сводя к минимуму побочные эффекты, связанные с минералокортикоидами.
Сигналинг глюкокортикоидов
Липофильные глюкокортикоиды путём диффузии проникают через цитоплазматические мембраны и связываются с находящимися в цитозоле глюкокортикоидными рецепторами. Данные рецепторы повсеместно экспрессируются ядросодержащими клетками организма. Человеческие рецепторы глюкокортикоидных гормонов кодируются геном NR3C1, содержащим 9 экзонов (см. доп. информацию). Подобно другим ядерным рецепторам (действующим в пределах клеточного ядра), рецептор глюкокортикоидов состоит из трёх доменов:
- первый, N-концевой домен, взаимодействует с корегуляторами и компонентами транскрипционного аппарата;
- второй, ДНК-связывающий домен, содержит два мотива «цинковых пальцев» для геномных взаимодействий;
- третий, лиганд-связывающий домен, напоминает гидрофобный карман и служит, собственно, для связывания лиганда [12].
Подвижный петлевой регион, который способствует димеризации рецептора, ядерной транслокации и связыванию с ДНК, располагается между ДНК-связывающим и лиганд-связывающим доменами [12]. Рецепторы глюкокортикоидов имеют множество сайтов для посттрансляционной модификации, включая сайты фосфорилирования, ацетилирования, убиквитинилирования и сумоилирования (SUMOylation), которые играют важную роль в ядерном импорте и экспорте, генной регуляции и деградации рецепторов [13].
Белки, синтезируемые на мРНК-копии гена NR3C1, неоднородны. Классическим рецептором, ассоциированным с транскрипционной регуляцией глюкокортикоидами, является GRα. GRβ, в противоположность GRα, представляет собой вариант сплайсинга, который не связывает лиганды, однако, как считается, является доминантным ингибитором GRα. Кроме того, есть упоминания и о прямой транскрипционной активности GRβ [13]. Накапливающиеся экспериментальные данные указывают на то, что воспаление оказывает влияние на профиль экспрессии той или иной изоформы рецептора глюкокортикоидов, а усиленная экспрессия GRβ была связана с устойчивостью к глюкокортикоидам при некоторых патологиях [14]. Далее спектр белков глюкокортикоидных рецепторов расширяется вследствие наличия восьми альтернативных сайтов инициации трансляции; формирующиеся изоформы по N-концевому домену рецептора глюкокортикоидов обладают выраженной транскрипционной активностью, способны к ядерно-цитоплазматическому транспорту и встречаются в клетках различных тканей [15]. Различия в экспрессии изоформ рецепторов глюкокортикоидов могут в какой-то степени объяснить клеточно-специфический ответ на глюкокортикоиды.
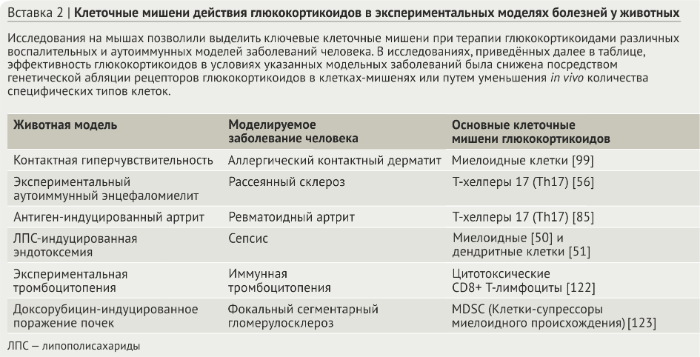
В отсутствие лиганда GRα локализованы в цитоплазме в виде мультипротеинового комплекса с белками теплового шока, иммунофиллинами и другими шаперонами, которые усиливают сродство рецептора к лиганду и предотвращают деградацию рецептора (рис. 2). По классическому пути связывание с лигандом индуцирует конформационное изменение рецептора, вследствие чего комплекс с шаперонами распадается, что в итоге делает возможным транспорт рецептора в ядро. Однако недавние исследования свидетельствуют о том, что шапероны также важны для переноса рецептора в ядро [16]. В ядре рецепторы глюкокортикоидов взаимодействуют с ДНК и другими белками, опосредуя развитие биологического ответа (рис. 2). Отсоединение лиганда ведёт к кальретикулин-опосредованному экспорту рецептора из ядра обратно в цитоплазму, где он снова переходит в комплексную форму, объединяясь с шаперонами и приобретая способность снова связаться с лигандом для запуска следующего цикла [17].
Как упоминалось выше, эндогенные глюкокортикоиды также обладают высоким сродством к рецепторам минералокортикоидов (кодируются геном NR3C2), регулирующих гены, продукты которых участвуют в реабсорбции натрия. По своему характеру экспрессия рецепторов минералокортикоидов более специфична по сравнению с рецепторами глюкокортикоидов; их высокая экспрессия встречается в протоках и канальцах почек, сердце и гиппокампе, а вот в лейкоцитах их биосинтез снижен. Концентрация глюкокортикоидов в крови обычно намного выше, чем эндогенного минералокортикоида альдостерона, но в чувствительных к минералокортикоидам тканях специфичность минералокортикоидных рецепторов к альдостерону обеспечивается ферментом 11βHSD2, который, инактивируя кортизол, препятствует занятию минералокортикоидных рецепторов глюкокортикоидами. Хотя информации о роли рецепторов минералокортикоидов в иммунитете мало, стоит отметить, что лейкоциты не экспрессируют 11βHSD2, что повышает вероятность того, что минералокортикоидные рецепторы лейкоцитов “не защищены” от блокирования эндогенными глюкокортикоидами.
Генная регуляция глюкокортикоидных рецепторов
Модуляцию функций иммунной системы посредством глюкокортикоидов, как правило, связывают с рецептор-опосредованными изменениями генной экспрессии. Варианты воздействия глюкокортикоидов на геном подразделяются на три основных механизма (рис. 2). Первый способ регуляции осуществляется за счёт действия рецепторов глюкокортикоидов как факторов транскрипции и задействует прямое связывание комплекса рецептор-лиганд с ДНК. Гомодимеры глюкокортикоидных рецепторов связываются на элементах глюкокортикоидной регуляции (GREs; имеют консенсусную последовательность GGAACAnnnTGTTCT, в которой «n» — любое основание) для усиления генной экспрессии [18,19]. Связанные с GRE рецепторы привлекают корегуляторы, в том числе коактиватор стероидных рецепторов (SRC1, steroid receptor coactivator 1, также известен как NCOA1), глюкокортикоид-рецептор взаимодействующий белок 1 (GRIP1, glucocorticoid receptorinteracting protein 1, также известен как NCOA2) и CREB-связывающий белок (CBP, CREB-binding protein, также известный CREBBP) и затем связывают активатор транскрипции BRG1 хроматин-ремодулирующего комплекса SWI/SNF, в результате чего образуется преинициирующий комплекс и запускается транскрипция.
Мономеры глюкокортикоидных рецепторов, наоборот, могут связываться с негативными GRE (nGRE; имеют консенсусную последовательность CTCC(n)0–2GGAGA) и рекрутируют корепрессоры, как то нуклеарный корепрессор (NCOR1) и SMRT (известен также как NCOR2), подавляя генную экспрессию [20]. Считалось, что GRE и nGRE являются компонентами промоторов генов, однако недавние полногеномные исследования показали, что большинство рецептор-связывающих сайтов располагаются во внутригенных областях и регионах, весьма удалённых от сайтов инициации транскрипции, что указывает на то, что глюкокортикоидные рецепторы могут оказывать регулирующее влияние на значительном расстоянии внутри генома [21]. Также полногеномные исследования показали, что имеется некоторое совпадение сайтов связывания глюкокортикоидных рецепторов среди различных тканей и типов клеток, что может свидетельствовать о том, что доступность хроматина является предпосылкой для взаимодействия рецепторов к глюкокортикоидам с ДНК [22]. Доступность GRE и nGRE, вероятно, способствует типоспецифическому воздействию глюкокортикоидов на клетки.
Второй способ глюкокортикоид-рецептор-опосредованной генной регуляции осуществляется путём физического взаимодействия глюкокортикоидного рецептора с другими факторами транскрипции (как бы «привязывает» их) без контакта с ДНК [23]. Такие белок-белковые взаимодействия изменяют способность партнёрствующего фактора транскрипции связываться с ДНК или рекрутировать корегуляторы и/или транскрипционный аппарат. Такое “привязывание” (tethering) стало объектом пристального внимания как один из механизмов глюкокортикоид-опосредованного ингибирования иммунного ответа, т.к. глюкокортикоидные рецепторы препятствуют действию ключевых провоспалительных факторов транскрипции, к примеру, NFκB (nuclear factor κB) и AP1 (activator protein 1), а также представителей семейств сигнальных трансдукторов и активаторов транскрипции (STAT), CCAAT/энхансер-связывающихся белков (C/EBP) и ядерных факторов активированных Т-клеток (NFAT) (вставка 3). Привязывание глюкокортикоидных рецепторов является не односторонним взаимодействием; многочисленные исследования показали, что это прикрепление оказывает влияние на способность рецепторов активировать гены-мишени [23].
Примечания:
RE — response element (регуляторные элементы, элементы отклика);
TM — transcriptional machinery (транскрипционный аппарат) [27].
Третий способ регуляции заключается во влиянии глюкокортикоидов на генную экспрессию посредством связывания глюкокортикоидных рецепторов с составными элементами, которые представляют собой последовательности ДНК, в составе которых присутствует как GRE, так и элемент отклика для отдельного фактора транскрипции [24]. И хотя обычно глюкокортикоидные рецепторы подавляют активность транскрипционного фактора как в составном элементе, так и в случае “привязывания”, тем не менее важно отметить, что связывание глюкокортикоидных рецепторов может усиливать активность партнёрского фактора транскрипции. К примеру, AP1 является частым партнёром рецепторов к глюкокортикоидам в составных элементах на протяжении всего генома [25], однако оказываемое рецепторами воздействие зависит компоновки субъединиц AP1; глюкокортикоидные рецепторы сильнее ингибируют активность гетеродимеров AP1, содержащих JUN и FOS, тогда как функциональная активность гомодимеров AP1, включающих в себя JUN–JUN, ограничивается глюкокортикоидами в меньшей степени и может быть даже усилена [26,27]. Аналогичным образом глюкокортикоидные рецепторы по-разному влияют на активность различных представителей семейства белков STAT [28] (вставка. 3).
Привлекательное, но противоречивое мнение гласит, что глюкокортикоиды оказывают иммуносупрессивное действие в основном путём связывания глюкокортикоидных рецепторов с такими факторами транскрипции, как NFκB и AP1, что снижает уровень экспрессии провоспалительных генов (трансрепрессия), тогда как нежелательные побочные эффекты возникают вследствие активации генов путём прямого связывания глюкокортикоидных рецепторов с GREs (трансактивация). Представление о том, что клинически полезные и вредные свойства глюкокортикоидов разделяются на молекулярном уровне, возникло в ходе наблюдения рецепторов глюкокортикоидов с димеризационными мутантами доменов, которые не вызывали экспрессии определённых генов, несущих GRE, но подавляли активность AP1 и NFκB [29]. Эти наблюдения подогревали фармацевтический поиск лигандов глюкокортикоидных рецепторов, которые бы специфически поддерживали трансрепрессивные свойства глюкокортикоидных рецепторов при минимальной индукции трансактивации [30]. На сегодняшний момент, однако, принято считать, что принцип трансактивации/трансрепрессии, вероятно, является упрощением влияния глюкокортикоидов на иммунные процессы, особенно в свете сообщений о том, что димеризационные мутанты рецепторов глюкокортикоидов сохраняют некоторую способность к активации гена транскрипции [31]. Более того, остается неясным то, сохраняют ли мономеры димеризационных мутантов рецепторов способность связывать nGRE и таким образом подавлять транскрипцию гена. Степень иммуномодуляции, осуществляемой с помощью глюкокортикоид-рецептор-опосредованной генной регуляции, в отличие от той, которая происходит путём ингибирования факторов транскрипции, до сих пор не ясна, но несколько генов, являющихся непосредственным объектом воздействия глюкокортикоидов, как обсуждается далее, оказывают сильное регулирующее воздействие на иммунный ответ.
Негеномные эффекты глюкокортикоидов
Тогда как геномные эффекты глюкокортикоидов проявляются обычно в течение нескольких часов, глюкокортикоиды в пределах от нескольких секунд до минут вызывают также биологические изменения, которые не являются результатом изменения транскрипции генов. Интеркаляция (встраивание) глюкокортикоидов в мембраны подразумевает под собой независимый от глюкокортикоидных рецепторов механизм для изменения катионного транспорта через цитоплазматические мембраны и для поддержания утечки протонов из митохондрий [32]. Как сообщается, глюкокортикоидные рецепторы, напротив, мешают цитоплазматическим сигнальным комплексам [33]. Кроме того, лиганд-зависимое разобщение молекулярных компонентов комплекса рецептор–шаперон влечёт за собой ослабление передачи сигнала [33]. Также есть сообщения о том, что лиганд-связанные глюкокортикоидные рецепторы транслоцируются в митохондрии, что приводит к инициации апоптоза [34].
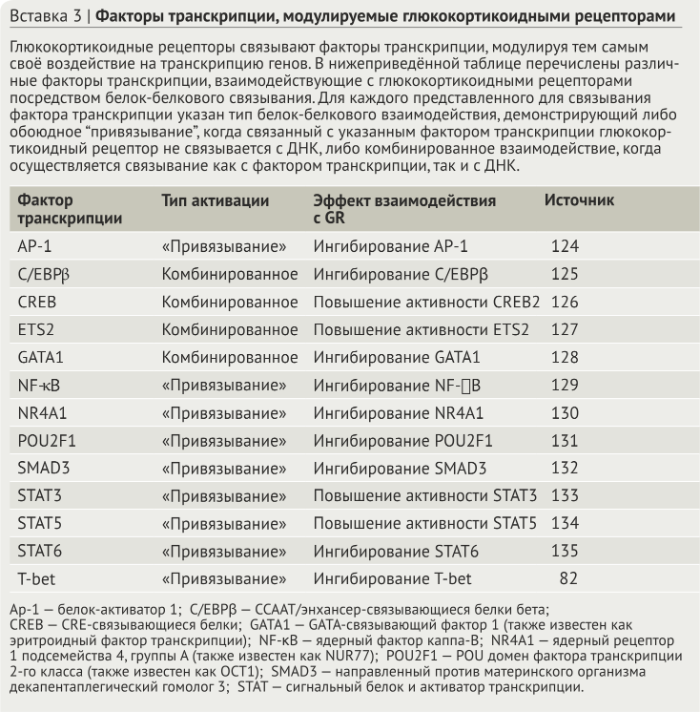
Регуляция воспаления
Глюкокортикоиды являются мощными регуляторами воспаления. Воспалительный ответ включает серию взаимосвязанных процессов, которые обычно можно разделить на три последовательные фазы: первая — фаза тревоги, в которой сигналы «опасности» вызывают высвобождение медиаторов воспаления; вторая — мобилизационная фаза, в которой лейкоциты проникают в поврежденный участок; и, наконец, фаза разрешения, в котором ткань очищается от клеточного мусора. Успешное прогрессирование и разрешение воспалительного ответа имеют решающее значение при заживлении ран. Как подробно описано ниже и на рис. 3, глюкокортикоиды регулируют процессы в каждой из этих фаз.
Роль глюкокортикоидов в фазе тревоги
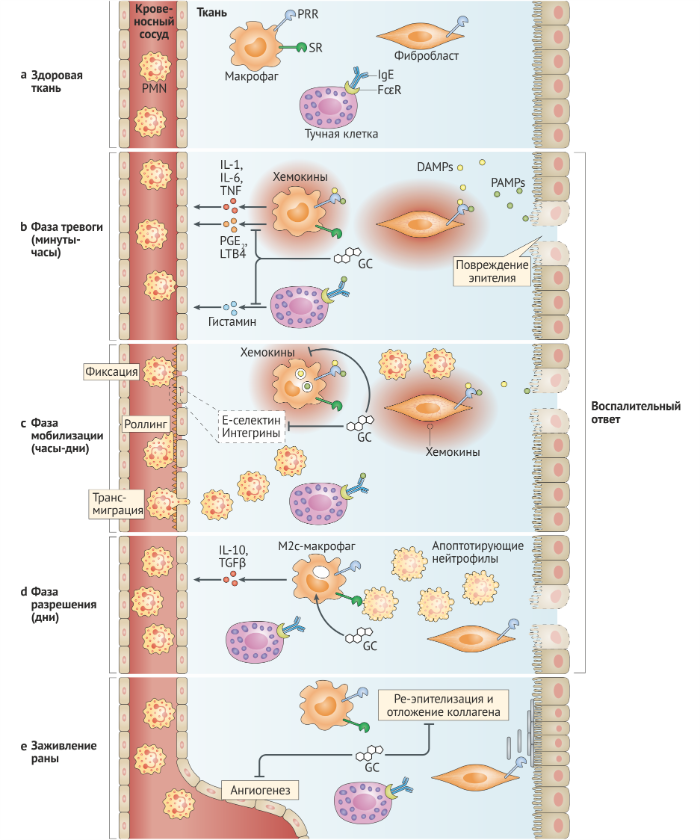
Фаза тревоги воспалительного ответа возникает при обнаружении патогенов и/или повреждения тканей. Резидентные клетки ткани экспрессируют трансмембранные и цитоплазматические белки, которые функционируют как сенсоры «опасности»; они включают в себя рецепторы распознавания паттернов (PRR, pattern recognition receptors), рецепторы комплемента и Fc-рецепторы (рис. 3a). PRR связывают патоген-ассоциированные молекулярные структуры (PAMP), которые являются эволюционно закреплёнными молекулярными мотивами от микробов и связанными с повреждением молекулярными структурами (DAMP), являющимися эндогенными клеточными продуктами, которые высвобождаются при повреждении тканей. После активации PRR тканевые макрофаги, тучные клетки и стромальные клетки секретируют воспалительные медиаторы, включая липидные медиаторы, вазоактивные амины и цитокины (рис. 3b). Глюкокортикоиды подавляют афферентные сигнальные пути от многих сенсоров, тем самым подавляя продукцию медиаторов воспаления. Например, передача сигналов TLR регулируется глюкокортикоидами на нескольких этапах сигнального пути рецептора (рис. 4). После передачи сигналов от TLR лигандный глюкокортикоидный рецептор связывает и ингибирует транскрипционные факторы, включая NF-κB, AP-1 и регуляторный фактор интерферона 3 (IRF3) (рис. 4).
b) В фазу тревоги патоген-ассоциированные молекулярные структуры (PAPM) и связанные с опасностью молекулярные структуры (DAMP) инициируют сигнал PRR, который включает образование медиаторов воспаления, таких как цитокины, простагландин E2 (PGE2) и лейкотриен B4 (LTB4). Антиген связывает FcεR-связанный IgE на тучных клетках, приводя к высвобждению гистамина. Глюкокортикоиды подавляют передачу сигнала через PRR, FcεRs и рецепторы цитокинов.
с) В фазу мобилизации медиаторы воспаления увеличивают количество адгезивных молекул, таких как Е-селектин, хемокины и интегрины, на поверхности эндотелия сосудов, чтобы привлечь лейкоциты, особенно полиморфноядерные лейкоциты (ПЯЛ), в ткань. Выходящие за пределы сосудов лейкоциты следуют градиенту концентрации хемокинов к очагу воспаления. Глюкокортикоиды ингибируют экспрессию Е-селектина, хемокинов и интегринов, чтобы уменьшить количество “прибывающих” лейкоцитов.
d) В фазу разрешения воспаления глюкокортикоиды способствуют дифференциации альтернативно активированных макрофагов “M2c”, которые очищают апоптические PMN и секретируют противовоспалительные факторы.
e) Воспаление завершается заживлением раны, которое характеризуется эпителизацией, отложением коллагена и ангиогенезом. Поскольку глюкокортикоиды ингибируют эти процессы, оптимальное заживление ран, вероятно, зависит от снижения их продукции.
IL, интерлейкин;
TGFβ, трансформирующий фактор роста β;
TNF, фактор некроза опухоли [27].
Глюкокортикоиды также активируют гены, кодирующие ингибиторы TLR, включая протеиновую фосфатазу 1 с двойной специфичностью (DUSP1), ослабляющую активность митоген-активированной протеинкиназы 1 (МАРК1), и IL-1 рецептор-ассоциированную киназу 3 (IRAK3), которая является негативным регулятором рецепторных сигналов TLR и IL-1 [35] (рис. 4). Другая цель глюкокортикоидов — ген NFKIA, который кодирует IκBα — мощный цитоплазматический ингибитор NF-κB [36], но полная картина физиологической роли этого гена в реализации эффектов глюкокортикоидов пока обсуждается [37]. Глюкокортикоиды также вызывают экспрессию индуцированного глюкокортикоидами белка лейциновой застёжки (GILZ, также известного как TSC22D3), который дополнительно подавляет активность NF-κB [38]. Примечательно, что воспалительные сигналы могут отменять некоторые ингибирующие эффекты глюкокортикоидов; в плазмацитоидных дендритных клетках (pDC) устойчивая передача сигнала через TLR7 и TLR9 нарушает способность глюкокортикоидного рецептора ингибировать активность NF-κB, что может привести к развитию аутоиммунной патологии [39].
Важно отметить, что многие сигнальные компоненты TLR, которые сопровождают глюкокортикоиды, особенно NF-κB и AP-1, разделяются сигнальными путями других сенсоров “опасности” (а также рецепторов цитокинов). Например, глюкокортикоиды подавляют передачу сигналов через рецепторы Fcε, тем самым ослабляя высвобождение гистамина тучными клетками [40] (рис. 3b). Блокируя общие «модули» передачи сигналов, глюкокортикоиды подавляют многочисленные сигнальные пути, которые участвуют в выявлении вредных факторов и распространении воспалительных сигналов.
Воспаление характеризуется местным расширением сосудов, повышением сосудистой проницаемости и выходом плазмы в ткань. В воспалённой ткани глюкокортикоиды взаимодействуют с макрофагами и подавляют синтез эйкозаноидов, которые являются липидными медиаторами (подобно простагландинам и лейкотриенам), вызывающими расширение сосудов и повышение их проницаемости (рис. 3b). Лигандный глюкокортикоидный рецептор активирует эксперссию аннексина А1, который препятствует катализируемому фосфолипазой A2 высвобождению арахидоновой кислоты [41] — промежуточного продукта жирной кислоты в биосинтезе эйкозаноидов. Далее глюкокортикоиды препятствуют образованию простагландинов, ингибируя циклооксигеназу 2 (COX-2, также известную как PTGS 2) через подавление NF-kB свойств GILZ [42]. Глюкокортикоиды уменьшают приток крови к месту воспаления путём повышения экспрессии ангиотензин-превращающего фермента и эндотелина, а также сенсибилизации эндотелиальных клеток к вазоконстрикторам и ингибирования продукции вазодилататоров, таких как брадикинин [43]. На модели у животных с острым повреждением лёгких Веторацци и др. доказали, что глюкокортикоиды сохраняют барьерную функцию эндотелина, регулируя экспрессию макрофагов сфингозинкиназы 1 (которая кодируется SPHK1), снижающей сосудистую проницаемость [44].
Сигналы об опасности, обнаруженные PRR, распространяются посредством индукции синтеза и высвобождения цитокинов. Цитокины связываются со своими рецепторами на поверхности соседних клеток для стимуляции биологических эффектов, которые часто включают экспрессию ещё большего количества цитокинов. Помимо вышесказанного, цитокиновые сигнальные сети усиливают и формируют воспалительный ответ. Глюкокортикоиды ингибируют экспрессию многих провоспалительных цитокинов, включая IL‑1α, IL‑1β, IL‑2, IL‑3, IL‑4, IL‑5, IL‑6, IL‑11, IL‑12, IL‑13, IL‑16, IL‑17, интерферон-γ (IFNγ), фактор некроза опухолей (TNF) и гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) (рис. 3b). Как было отмечено выше, опосредованное глюкокортикоидами ослабление экспрессии цитокинов происходит после реализации сигнала от сенсора “опасности” и цитокинового рецептора [45]. Кроме того, nGRE были найдены в некоторых генах цитокинов, включая IL1B (который кодирует IL‑1β), IL6 и TSLP (который кодирует тимусный стромальный лимфопоэтин) [20]. Глюкокортикоиды также регулируют продукцию цитокинов на посттранскрипционном уровне. Опосредованная глюкокортикоидами рецепторная регуляция тристетрапролина (также известного как ZFP36) уменьшает период полураспада мРНК TNF [46]. В клетках, чувствительных к цитокину, глюкокортикоиды ослабляют передачу сигнала от цитокинового рецептора, проходя через белок-белковые взаимодействия между лигандным рецептором глюкокортикоидов и провоспалительными факторами транскрипции (например, члены семейства STAT и NF-kB), а также благодаря активации супрессора сигналинга цитокинов 1 (SOCS1), ингибитора Янус-киназы (JAK) — сигнала STAT [47,48]. Важность передачи сигналов от глюкокортикоидного рецептора при снижении продукции цитокинов наглядно иллюстрируется на примере мышей с условной абляцией глюкокортикоидных рецепторов в макрофагах или дендритных клетках (ДК). Эти мыши продуцируют повышенные уровни IL-1, IL-6, TNF и IL-12 и имеют более высокую смертность во время экспериментально вызванного сепсиса, чем животные с диким фенотипом [49‒51].
Роль глюкокортикоидов в фазе мобилизации
Воспалительные медиаторы, продуцируемые в фазе тревоги, вызывают экспрессию молекул адгезии и хемоаттрактантов, которые “притягивают” лейкоциты из проксимальных кровеносных сосудов в очаг воспаления. Фаза мобилизации имеет решающее значение для очистки от патогенных микроорганизмов и клеточного мусора. Эндотелиальная экспрессия E-селектина инициирует адгезию циркулирующих нейтрофилов и моноцитов к стенкам кровеносных сосудов, что облегчает взаимодействие между хемокинами, поступающими от участков воспаления и хемокиновых рецепторов на лейкоцитах (рис. 3c). Взаимодействие хемокина с его рецепторами способствует перемещению лейкоцитов по эндотелию и связыванию интегринов с лигандами интегринов, что приводит к устойчивой адгезии и трансмиграции лейкоцитов через стенку кровеносного сосуда [52] (рис. 3c). Затем вышедшие из сосудов лейкоциты мигрируют через ткань в очаги воспаления, следуя градиенту хемокинов.
Глюкокортикоиды ослабляют трансмиграцию лейкоцитов, ингибируя эндотелиальную транскрипцию SELE (который кодирует E-селектин), а также лиганды интегринов ICAM1 (молекула межклеточной адгезии 1) и VCAM1 (молекула адгезии сосудистых клеток 1) [53,54]. Глюкокортикоиды также подавляют синтез нескольких хемокинов и хемоаттрактантов, включая IL-8 (также известный как CXCL8), IL-16, CC-хемокиновый лиганд 2 (CCL2), CCL3, CCL5, CCL11, CCL24 и CCL26, тем самым обуславливая миграцию лейкоцитов. Недавние исследования показали, что непосредственное связывание глюкокортикоидного рецептора с транскриптами мРНК, кодирующей хемокины, включая CCL2 и CCL7, способствует их распаду [55]. Глюкокортикоиды также снижают экспрессию молекул адгезии лейкоцитов, включая CD44 и ассоциированный с функцией лимфоцитов интегрин 1 (LFA1) и очень поздний антиген 4 (VLA4) [56]. Глюкокортикоид-опосредованная индукция аннексина А1 также препятствует рекрутированию лейкоцитов [57]. На модели экспериментального аутоиммунного энцефаломиелита и рассеянного склероза у животных глюкокортикоиды подавляли инфильтрацию Т-лимфоцитами центральной нервной системы [56].
Роль глюкокортикоидов в фазе разрешения
Поскольку патогенные микроорганизмы и вредные агенты удаляются из участков воспаления, передача сигналов PRR и производство медиаторов воспаления уменьшается. Однако воспаление полностью прекращается, когда активируется программа иммунорегуляции, при которой важную роль играют глюкокортикоиды — наряду с липоксинами, резолвинами, коллективинами и протектинами [58]. Важными процессами на стадии разрешения являются очистка от клеточного мусора и производство противовоспалительных факторов. Глюкокортикоиды инициируют программы генов в моноцитах и макрофагах, которые способствуют фагоцитозу апоптотировавших клеток и остального клеточного мусора [59‒61].
Опосредованный глюкокортикоидами контроль макрофагов привлёк достаточно внимания, чтобы классифицировать их как подтип «M2c» альтернативно активированных макрофагов; макрофаги в этом состоянии характеризуются высокой экспрессией скавенджер рецепторов (scavenger receptors — CD163 (также известный как M130), CD206 (также известный как MRC1), тирозин-протеинкиназы MER (MERTK)) и секрецией противовоспалительных цитокинов, трансформирующего фактор роста β (TGFβ) и IL-10 (рис. 3d) [62]. Другая функция глюкокортикоидов в фазе разрешения заключается в сенсибилизации клеток к другим про-разрешающим факторам посредством апрегуляции рецептора липоксина A4 [63]. За разрешением воспалительного ответа следует заживление ран, которое восстанавливает целостность и функцию тканей. Глюкокортикоиды подавляют многие процессы заживления ран, включая отложение коллагена, эпителизацию и ангиогенез; ослабление активности глюкокортикоидов может быть важным компонентом перехода от воспаления к заживлению ран.
Активированный GR повышает транскрипцию генов, кодирующих ингибиторы сигналинга TLR, такие как IL-1 рецептор-ассоциированная киназа 3 (IRAK3), фосфатаза двойной специфичности второго типа (DUSP1), ингибитор ядерного фактора kB (IkB) и глюкокортикоид-индуцированная лейциновая застежка-молния (GILZ). GR также снижает активность провоспалительных транскрипционных факторов, таких как белок-активатор 1 (AP-1), ядерный фактор-kB (NF-kB) и интерферон-регулирующий фактор 3 (IRF3) посредством белок-белковых взаимодействий.
IKKε, IκB kinase-ε;
JNK, JUN N-концевая киназа;
MKK, митоген-активированная протеинкиназа (также известная как MAP2K);
MYD88, белок первичного ответа миелоидной дифференцировки-88;
TAK1, трансформирующий фактор роста β-активированной киназы 1 (также известен как MAP3K7);
TBK1, TANK-связывающая киназа 1;
TM, транскрипционный механизм;
TRIF, содержащий TIR-домен адаптер белковой индукции IFNβ (также известный как TICAM1) [27].
Регуляция клеточной гибели глюкокортикоидами
Глюкокортикоиды оказывают сильные регулирующие эффекты на клеточный иммунитет и влияют на развитие, активацию и дифференцировку Т-лимфоцитов. Определить роль глюкокортикоидов в развитии тимоцитов довольно затруднительно. Существуют противоречивые данные о рецептор-опосредованной регуляции тимопоэза глюкокортикоидами. Тимоциты крайне чувствительны к глюкокортикоидным сигналам клеточной гибели in vitro и in vivo; более того, резекция надпочечников приводит к гиперплазии тимуса — это может означать, что недостаточность глюкокортикоидов способствует чрезмерному размножению популяций тимоцитов. Эти наблюдения являются основным доказательством длительно существующей гипотезы о негативной регуляции тимопоэза эндогенными глюкокортикоидами. Однако, у химерных животных [64] с недостаточностью глюкокортикоидных рецепторов в фетальной печени не проявляется фенотип с тимомегалией, характерный для мышей с резекцией надпочечников. Более того, у мышей с Т-специфичной абляцией глюкокортикоидного рецептора все ещё сохранялась гиперплазия тимуса, вызванная резекцией надпочечников. Это говорит о том, что глюкокортикоидная недостаточность усиливает экспансию тимоцитов независимо от сигналинга глюкокортикоидного рецептора (или его недостатка) в этих клетках [65].
Проблема изучения роли глюкокортикоидов в тимопоэзе усложняется данными о наличии как положительной, так и отрицательной роли глюкокортикоидов в выживании тимоцитов. В соответствии с гипотезой “взаимного антагонизма”, взаимодействие между глюкокортикоидным и Т-клеточным рецептором (TCR) изменяет порог между положительной и отрицательной селекцией во время развития тимоцитов. В поддержку этой гипотезы существует недавнее исследование на линии мышей с Т-специфической делецией глюкокортикоидного рецептора. В этом исследовании мутация уменьшила количество клеток тимуса, изменила TCR-репертуар и ослабила Т-специфический ответ [66]. Хотя стрессовые и фармакологические уровни глюкокортикоидов обладают исключительно негативными эффектами на выживаемость тимоцитов, роль эндогенных глюкокортикоидов в стабильной регуляции тимопоэза остаётся неясной.
Влияние глюкокортикоидов на лимфоциты обычно используется для лечения некоторых злокачественных заболеваний системы крови. Несмотря на то, что глюкокортикоиды были одними из первых описанных стимуляторов гибели лимфоцитов, молекулярные механизмы, инициирующие апоптоз после активации глюкокортикоидных рецепторов, до сих пор не ясны. Так, есть некоторая ирония в том, что глюкокортикоиды нередко используют в качестве контрольных препаратов для индукции апоптоза в исследованиях новейших проапоптотических и противоапоптотических факторов. Механизм апоптоза, опосредованного глюкокортикоидами, всесторонне изучен [33,67], и важно отметить, что механизмы активированного глюкокортикоидами апоптоза различаются по условиям активации, типам лимфоцитов и даже по стадии дифференцировки [68].
По-видимому, в программах апоптоза, вызванных глюкокортикоидами, имеется значительная избыточность, потому что лимфоциты от многих мышей с одним нокаутированным геном (например, нокаутированы гены, кодирующие индивидуальные члены семейства каспаз) продолжают быть чувствительными к сигналам гибели, опосредованным глюкокортикоидными рецепторами. Считается, что апоптоз, опосредованный глюкокортикоидными рецепторами, требует активации генов, хотя в последних исследованиях были обнаружены внегеномные механизмы, например, перемещение глюкокортикоидного рецептора на мембрану митохондрий [33]. N-концевые изоформы глюкокортикоидного рецептора обладают неодинаковой способностью вызывать апоптоз, поэтому профиль изоформ глюкокортикоидного рецептора клетки, вероятно, может влиять на чувствительность к глюкокортикоид-индуцированной гибели [69]. Наконец, стоит упомянуть, что лимфоциты — не единственные клетки, чувствительные к проапоптотическим сигналам глюкокортикоидов. Дендритные клетки [70], эозинофилы [71] и остеоциты [72] также подвергаются апоптозу в ответ на воздействие глюкокортикоидов.
Эффекты глюкокортикоидов, оказываемые на активацию Т-лимфоцитов
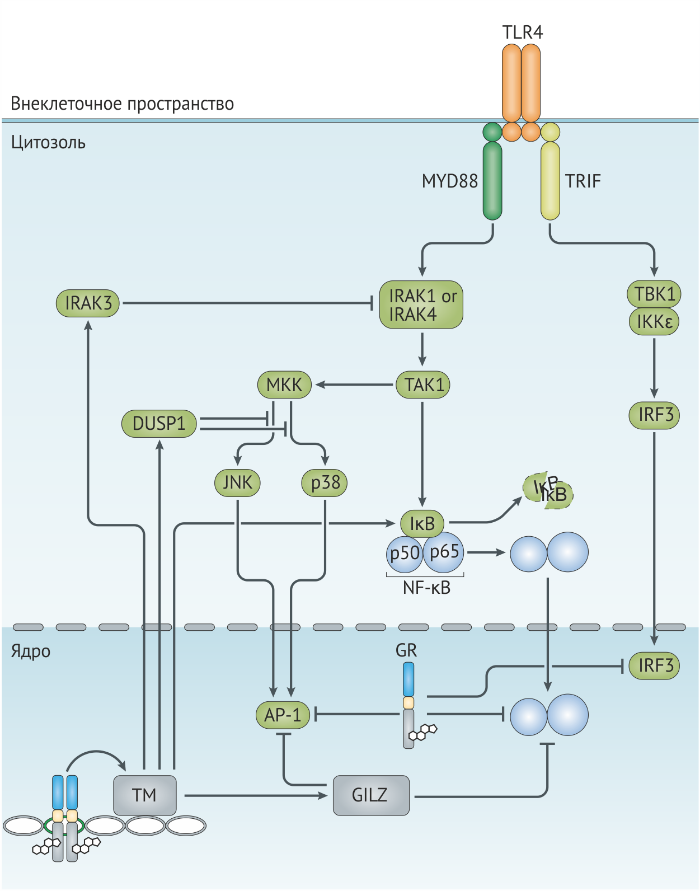
Ключевой компонент клеточного иммунитета — это TCR-опосредованная активация антигенспецифичных Т-лимфоцитов. Антигенпрезентирующие клетки (АПК) демонстрируют белковый антиген, упакованный в молекулы MHC I или II класса для активации CD8+ или CD4+ Т-лимфоцитов. Дендритные клетки являются профессиональными АПК, объединяющими антигены и сигналы от врождённой иммунной системы для стимуляции Т-хелперного ответа, который является одной из мишеней глюкокортикоидов. Эффекты глюкокортикоидов были изучены на различных типах дендритных клеток, включая тканевые, мигрирующие и плазмоцитоидные дендритные клетки. Были сделаны выводы, что глюкокортикоиды снижают активность дендритных клеток путём:
- ингибирования их созревания;
- посредством нисходящей регуляции экспрессии молекул МНС II, липид-представляющих молекул CD1a, ко-стимулирующих молекул (например, CD80 и CD86) и провоспалительных цитокинов (таких, как IL-12 и TNF);
- стимуляции экспрессии противовоспалительных цитокинов, таких как IL-10 (рис. 5а).
Интересно, что воздействие глюкокортикоидов во время перехода моноцита в дендритную форму увеличивает способность дендритных клеток захватывать антиген, но снижает их антигенпрезентирующую функцию, что привело к гипотезе о стимуяции глюкокортикоидами дифференцировки “толерогенных” дендритных клеток [74]. Более того, и у грызунов, и у людей фармакологические дозы глюкокортикоидов снижают количество дендритных клеток, что может быть результатом запуска апоптоза и перераспределения в тканях.
Глюкокортикоиды также могут тормозить активацию Т-лимфоцитов, вмешиваясь в Т-клеточный сигналинг. Механизмы, вовлечённые в глюкокортикоид-опосредованное снижение TCR-сигналинга, включают нисходящую регуляцию FOS и нарушение активности AP-1, NF-kB и NFAT [77] (рис. 5а). Недавнее исследование показывает, что глюкокортикоиды также могут снижать частоту экспрессии некоторых киназ, вовлечённых в TCR-сигналинг, например, ITK, TXK и LCK [78]. Внегеномное действие глюкокортикоидных рецепторов вовлечено в ингибирование TCR-сигналинга путём снижения активности киназ LCK и FYN [79] (рис. 5а). Ослабление TCR-сигналинга, опосредованное рецепторами глюкокортикоидов, приводит к снижению пролиферативного ответа и уменьшению продукции цитокинов, включая снижение секреции IL-2.
Хотя суммарный эффект глюкокортикоидов уменьшает активность Т-лимфоцитов, некоторые исследования говорят о том, что глюкокортикоиды преимущественно подавляют реакцию Th1- и Th17-лимфоцитов, при этом сохраняя, а иногда даже стимулируя функцию Th2-лимфоцитов и регуляторных Т-клеток (Treg) (рис. 5b). Механизмы предполагаемой глюкокортикоид-индуцированной Th2-поляризации включают down-регуляцию антигенпрезентирующими клетками продукции цитокина IL-12, стимулирующего Th1-клетки, снижение экспрессии рецептора к IL-12 на мембране Т-лимфоцитов, ингибирование мастер-фактора транскрипции Th1-лимфоцитов T-bet (TH1 cell master transcription factor Tbet) и повышение продукции Th2-лимфоцитами классических цитокинов Th2-типа (IL-4, IL-10 и IL-13) [81,82].
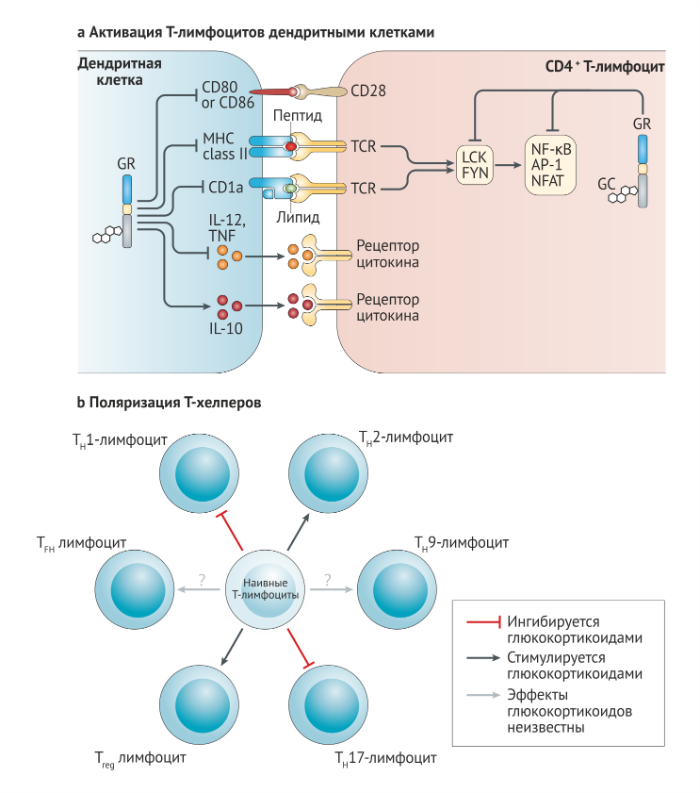
b. Глюкокортикоиды влияют на поляризацию Т-хелперов, направляя её в сторону Th2-лимфоцитов и регуляторных Т-клеток, а не в сторону Th1- и Th17-лимфоцитов. Для понимания эффектов глюкокортикоидов на дифференцировку Th9-клеток и фолликулярных Т-хелперов необходимы дальнейшие исследования.
AP-1, белок-активатор 1;
GR, глюкокортикоидный рецептор;
IL, интерлейкин;
NFAT, ядерный фактор активированных Т-лимфоцитов;
NF-kB, ядерный фактор-kB;
TNF, фактор некроза опухолей [27].
Свидетельства влияния глюкокортикоидов на поляризацию Th17-лимфоцитов исходят из репрессивных эффектов GILZ на продукцию Th17-стимулирующих факторов (таких, как IL-1, IL-6 и IL-23) дендритными клетками и на экспрессию генов, вовлечённых в дифференцировку и активность Th17-клеток (а именно IL-17A, рецептора к IL-23, RORγt, STAT3, BATF и IRF4) [83,84]. Исследования на мышах с Т-специфической делецией в гене глюкокортикоидного рецептора выявили, что Th17-лимфоциты являются ключевыми мишенями для глюкокортикоидной терапии на животных моделях при рассеянном склерозе и ревматоидном артрите [56,85]. Эти открытия могут хотя бы частично объяснить способность глюкокортикоидов стимулировать аутоиммунный и воспалительный ответ, включая псориаз и спондилоартропатию.
Исследования на мышах и людях также связывают глюкокортикоиды с активацией дифференцировки и активности Treg. Лечение глюкокортикоидами основано на повышении в циркулирующей крови и/или воспалительных тканях Treg. Этот эффект, вероятно, опосредован индуцированной глюкокортикоидными рецепторами up-регуляцией регуляторного Т-клеточного мастер-фактора транскрипции семейства forkhead box P3 (FOXP3), который, вероятно, активируется в результате up-регуляции GILZ.
Глюкокортикоиды и гуморальный иммунитет
Наше понимание влияния глюкокортикоидов на гуморальный иммунитет ограниченно. Тем не менее глюкокортикоиды эффективны в лечении аутоиммуных заболеваний, развивающихся при участии антител, таких как ревматоидный артрит. Более того, недавние исследования показали, что В-лимфоциты обладают мощными иммунорегуляторными функциями, особенно через экспрессию ИЛ-10 (ген ИЛ-10 известен как мишень глюкокортикоидов в макрофагах), что, возможно, приведёт к новому взгляду на влияние глюкокортикоидов на B-лимфоциты.
Влияние на развитие и выживание В-лимфоцитов
В некотором роде действие глюкокортикоидов на тимопоэз отражается и на В-лимфопоэзе. Юные В-лимфоциты более чувствительны ко глюкокортикоид-индуцированному апоптозу, чем зрелые В-лимфоциты [88,89], и адреналэктомия или введение антагониста рецепторов глюкокортикоидов мифепристона приводят к росту популяции юных В-лимфоцитов в костном мозге [89]. В-лимфоциты экспрессируют рецепторы глюкокортикоидов в процессе созревания [90], и эти рецепторы оказывают влияние на несколько факторов транскрипции, исходящих от сигнальных путей рецепторов В-лимфоцитов (например AP1, NFκB и NFAT), что повышает возможности глюкокортикоидов оказывать влияние на селекцию В-лимфоцитов.
На наш взгляд, механизмы взаимодействия между сигнальными путями рецепторов глюкокортикоидов и рецепторов В-лимфоцитов (BCR) пока недостаточно изучены. Намёки на прямое влияние глюкокортикоидов на функции В-лимфоцитов можно увидеть в исследованиях мышей с отсутствием гена-мишени глюкокортикоидов Gilz. В ответ на стимуляцию BCR клетки с отсутствующим Gilz демонстрируют повышенный пролиферативный ответ, и с возрастом у Gilz-нокаутных мышей развивается волчаночноподобный синдром [91]. Мыши с отсутствием Gilz в В-лимфоцитах не демонстрируют аутоиммунный фенотип глобального (т.е. во всех клетках, прим. перев.) дефицита Gilz, что показывает выходящую за пределы В-лимфоцитов роль GILZ в продукции аутоантител; однако, мыши с избирательным дефицитом экспрессии GILZ в В-лимфоцитах демонстрируют системное увеличение числа В-лимфоцитов, что подтверждает специфичную роль сигналинга рецепторов глюкокортикоидов в выживании и/или гомеостазе В-лимфоцитов.
Влияние на продукцию антител
Пока не удалось достичь общего согласия насчёт влияния глюкокортикоидов на гуморальный иммунитет, хотя в общем фармакологическое лечение использованием глюкокортикоидов связано со снижением концентрации иммуноглобулинов в крови. Лишь в нескольких исследованиях были рассмотрены последствия долгосрочного лечения глюкокортикоидами при вакцинации людей и было сообщено о небольшом воздействии (если таковое имелось) на титры антител. Тем не менее некоторое внимание получило влияние глюкокортикоидов на продукцию IgE, и некоторые клинические и in vitro исследования подтверждают, что глюкокортикоиды могут при определённых условиях способствовать продукции этого изотипа иммуноглобулинов. В исследовании пациентов с астмой, получавших терапию глюкокортикоидами, концентрации IgE в сыворотке возрастали, в то время как концентрации других иммуноглобулинов не изменялись или уменьшались [93,94]. Предлагаемые механизмы глюкокортикоид-опосредованного повышения продукции IgE включают прямое влияние на B-клетки путём переключения класса изотипов совместно с ИЛ-4 [95], а также посредством непрямых эффектов на В-лимфоциты через действие глюкокортикоидов на Т-лимфоциты и моноциты [96–98].
Эффективность глюкокортикоидов в лечении астмы и аллергий, однако, подтверждает, что глюкокортикоид-опосредованное повышение продукции IgE (так же, как и активности Th2-лимфоцитов) не способствует усугублению патологии при этих заболеваниях. Действительно, глюкокортикоиды оказывают значительные эффекты на миелоидные клетки и ослабляют заболевание в исследованиях аллергического дерматита на животных [99]. В свете клинических доказательств способности глюкокортикоидов снижать концентрации некоторых не-Е иммуноглобулинов в крови становится непонятным, каким образом эндогенные глюкокортикоиды в то же время являются позитивными регуляторами гуморального (посредством антител) ответа на иммунизацию. Например, у крыс с адреналэктомией наблюдаетя ослабленная секреция IgM и IgG в ответ на введение Т-зависимого антигена гемоцианин фиссуреллы (KLH, keyhole limpet haemocyanin; фиссурелла — брюхоногий моллюск, прим. перев.) [100,101]. Активация Т-опосредованного гуморального ответа включает экспрессию рецепторов глюкокортикоидов во многих типах клеток, включая ДК, CD4+ T-лимфоциты и B-лимфоциты. Определение глюкокортикоид-чувствительных клеток и процессов, которые стимулируют ответную секрецию антител, предоставит важную информацию об эндокринной регуляции гуморального иммунитета. В следующем разделе мы подробно рассмотрим загадочную природу глюкокортикоидов как усилителей иммунных реакций.
Иммуностимулирующие эффекты глюкокортикоидов
Этот обзор, как и большинство литературы о глюкокортикоидах, опубликованной за последние 60 лет, сфокусирован на иммуносупрессивных функциях глюкокортикоидов. Однако, существует значительное количество работ, показывающих, что глюкокортикоиды также усиливают воспаление и иммунитет. В некоторых исследованиях потенциирующие — как противоположность супрессивным — эффекты глюкокортикоидов связаны с дозой. Например, в макрофагах, активированных липополисахаридом (LPS) и интерфероном γ (IFNγ), высокие дозы кортикостерона ингибируют транскрипцию генов воспаления, в то время как низкие дозы кортикостерона усиливают экспрессию генов, связанных с воспалением [102]. Временное отношение между вредным стимулом и воздействием глюкокортикоидов может также влиять на направленность эффектов глюкокортикоидов при воспалении. У крыс экспрессия провоспалительных цитокинов усиливается, если глюкокортикоиды вводятся до воздействия ЛПС, но снижается, если глюкокортикоиды вводятся после ЛПС [103]. Похожим образом введение низких доз кортикостерона перед воздействием усиливает воспаление на модели реакции замедленной гиперчувствительности у крыс, в то время как высокие дозы или длительное введение глюкокортикоидов ослабляет иммунный ответ при ГЗТ [104].
Исследования полногеномной экспрессии подвергшихся воздействию глюкокортикоидов клеток принесли удивительные результаты и подтвердили, что глюкокортикоиды в значительной степени сдерживают или усиливают генные пути, связанные с врождённым иммунитетом, но избирательно подавляют пути, вовлечённые в адаптивный иммунитет (рис. 6а). Например, обработка моноцитов глюкокортикоидами приводит к up-регуляции нескольких PRR, скавенджер-рецепторов, рецепторов цитокинов и хемокинов, компонентов комплемента и других факторов, вовлечённых во врождённый иммунитет, но подавляет экспрессию генов, кодирующих компоненты TCR, а также генов, вовлечённых в активацию Т-лимфоцитов, включая гены МНС класса II и костимуляторынх молекул [1] (рис. 6а). Селективная стимуляция экспрессии генов врождённого иммунитета глюкокортикоидами также была показана на макрофагах человека [105].
Подсказки о молекулярных механизмах, лежащих в основе иммунопотенцирующих эффектов глюкокортикоидов, могут быть найдены среди генов-мишеней глюкокортикоидных рецепторов. Глюкокортикоиды стимулируют экспрессию TLR2, TLR4, компонент инфламмасом NOD, LRR и пуриновый домен-содержащего белка 3 (NACHT, LRR and PYD domains-containing protein 3, NLRP3) и пуринергического рецептора P2Y2R, который может сенситизировать клетки к PAMP и DAMP [106]. Интересно, что, хотя хорошо известна способность глюкокортикоидов подавлять секрецию цитокинов, также сообщается, что они увеличивают экспрессию некоторых рецепторов цитокинов, включая рецепторы к TNF, IL1, IL6 и IFNγ [107]. Резкое увеличение концентрации циркулирующих глюкокортикоидов, вызванное физиологическим стрессом, может выступать в качестве системы предупреждения и сенситизировать клетки к DAMP, PAMP и воспалительным цитокинам. Таким образом, глюкокортикоиды (наряду с катехоламинами) являются важными участниками изменения функции иммунитета, связанного с хроническим стрессом [108,109].
Единая модель
Как обсуждалось выше, у глюкокортикоидов показана способность как усиливать, так и подавлять функции иммунной системы. Чтобы объяснить этот очевидный парадокс действия глюкокортикоидов, Манк и Нерей Фейеш Тот предложили, что физиология действия глюкокортикоидов соответствует двухфазной кривой зависимости доза–реакция так, что глюкокортикоиды оказывают “разрешающие” (то есть, иммуностимулирующие) эффекты в низких концентрациях и подавляющие эффекты в высоких концентрациях [110,111].
Двухфазное действие глюкокортикоидов также наблюдается в других физиологических контекстах, включающих нейрональную пластичность [112]. Вигерс и Ройль [107] в дальнейшем предположили, что сеть клеточных эффектов конкурентной глюкокортикоид-опосредованной up-регуляции цитокиновых рецепторов и down-регуляции соответствующих цитокинов является биологическим ответом с более быстрым началом, но короткой продолжительностью. Мы предлагаем единую модель глюкокортикоид-опосредованной регуляции иммунного ответа, которая объединяет гипотезу двухфазного механизма доза–ответ, предложенную Манком и Фейеш Тотом [110], и кинетические эффекты разрозненной регуляции цитокинов и их рецепторов, описанной Вигерсом и Ройлем [107]. Мы предполагаем, что в отсутствие воспаления базальные уровни сигнальных путей рецепторов глюкокортикоидов, которые управляются циркадными и ультрадианными ритмами продукции глюкокортикоидов, сенситизируют клетки к повреждающим стимулам путём усиления экспрессии PRR, рецепторов цитокинов и факторов комплемента, что позволяет быстро обнаруживать PAMP и DAMP и способствовать индукции воспалительного ответа в ответ на повреждение ткани. Во время воспаления, однако, вызванные стрессом (или фармакологически) концентрации глюкокортикоидов сдерживают иммунный ответ в первую очередь путём ограничения распространения сигналов PRR, Fc-рецепторов и сигналов цитокинов, сокращая таким образом продолжительность иммунного ответа (рис. 6b).
Хотя это, вероятно, будет сильным упрощением действия глюкокортикоидов, мы предлагаем эту модель как основу для будущих исследований. Понимание механизмов и состояний, при которых глюкокортикоиды изменяют иммунный ответ — позитивно и негативно, возможно, принесёт новые идеи для оптимизации терапии глюкокортикоидами.
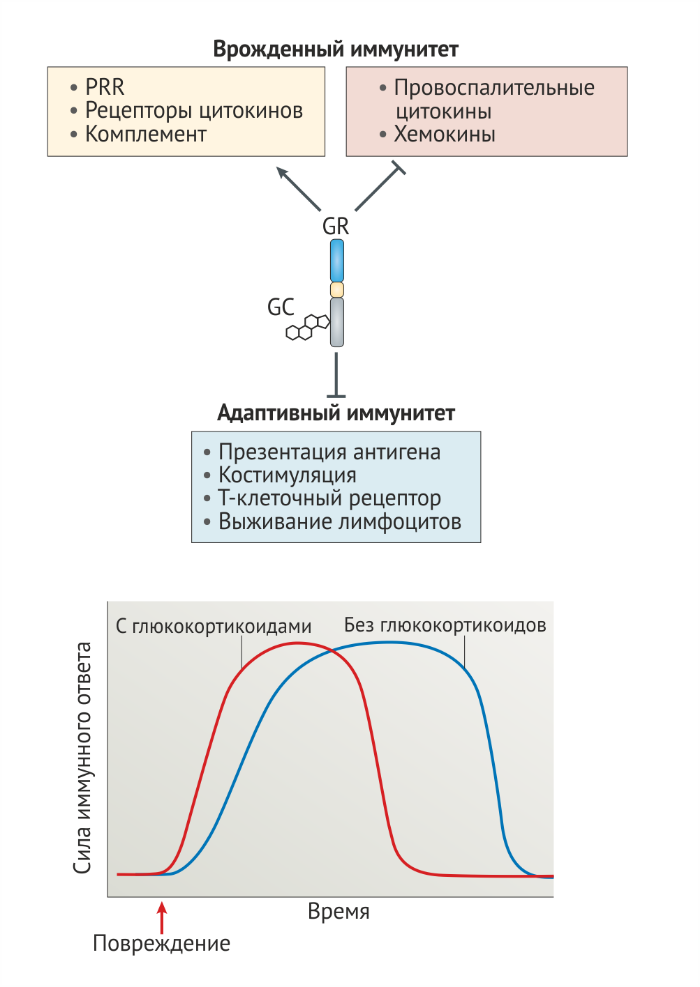
b) Гипотетическая линия времени для иммунного ответа, происходящего в присутствии (красная линия) и в отсутствии (синяя линия) глюкокортикоидов. Мы считаем, что глюкокортикоиды регулируют иммунную систему по двухфазному механизму так, что малые дозы способствуют экспрессии генов врождённого иммунитета и быстрому ответу на повреждение, но стрессовые и/или фармакологические концентрации подавляют передачу сигнала через иммунные рецепторы. Согласно этой гипотезе, животные с достаточным количеством глюкокортикоидов (красная линия) быстро реагируют на патогены и повреждение тканей, но эти реакции имеют контролируемую продолжительность. У животных с дефицитом глюкокортикоидов (синяя линия), однако, пониженная экспрессия PRR и рецепторов цитокинов приводит к замедлению иммунного ответа. Кроме того, в отсутствие глюкокортикоид-опосредованной супрессии сигнальных путей иммунных рецепторов продолжительность иммунного ответа увеличивается [27].
Заключение и перспективы
Клиническая эффективность глюкокортикоидов в лечении воспалительных и аутоиммунных заболеваний очевидна, хотя молекулярные механизмы, лежащие в основе иммунорегуляторных эффектов глюкокортикоидов, всё ещё изучаются. Существует большой разрыв между нашим пониманием глюкокортикоид-опосредованной регуляции иммунитета, особенно в отношении специфических функций клеточных линий и специфичных функций при заболеваниях, и возможными эффектами, зависящими от пола. Более того, побочные эффекты глюкокортикоидов широко варьируются (вставка 1); для достижения «святого грааля» безопасной терапии глюкокортикоидами необходимо более глубокое понимание являющихся общими для физиологических систем эффектов глюкокортикоидов, а также их уникальных эффектов в отношении иммунной системы.
Дополнительная информация
GeneCards entry for NR3C1: http://www.genecards.org/cgibin/carddisp.pl?gene=NR3C1
Nuclear Receptor Signaling Atlas entry for NR3C1: https://www.nursa.org/nursa/index.jsf
Online Mendelian Inheritance in Man entry for NR3C1: http://www.omim.org/entry/138040
The British Pharmacological Society and the International Union of Basic and Clinical Pharmacology (IUPHAR) Guide to PHARMACOLOGY entry for NR3C1: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=625
