
Фенилкетонурия

«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше...
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты. Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ. В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин. В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта. Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Итак…
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
Наиболее часто (~ 97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно. Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
В остальных ~ 2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».
«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* |
Уровень фенилаланина в крови, мкмоль/л |
Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) |
120–600 |
2–10 |
| Умеренная (мягкая, средняя) |
600–1200 |
10–20 |
| Классическая (тяжелая) |
>1200 |
>20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название |
Причинный фермент |
| ФАГ*-зависимая ФКУ** |
Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) |
6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B |
Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) |
Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D |
Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит |
Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях. У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Патогенез
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина. Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести. Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов). Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина.
Также на количество медиаторов ЦНС влияет и само количество фенилаланина. Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.
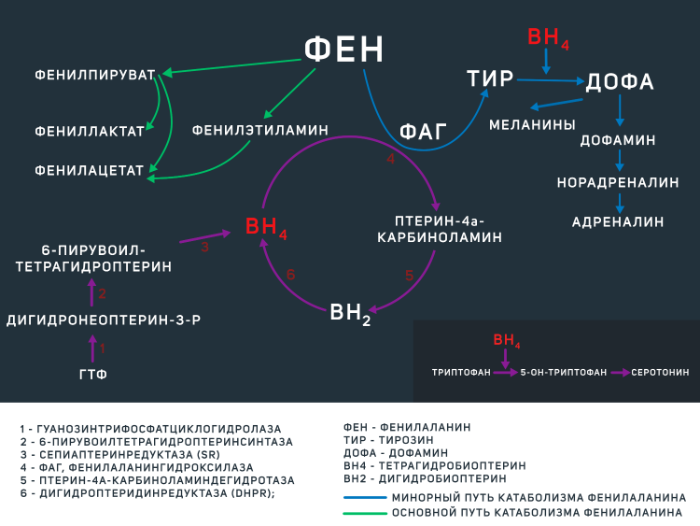
Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным». Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии. Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии. При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин. Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
Лечение
Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка = ~ 50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму.
Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока. - Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
Источники:
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] - Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] - М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend
