
Синдром Ретта

Синдром Ретта — крайне редкое наследственное заболевание, которое возникает у девочек преимущественно в раннем возрасте, характеризующееся регрессом психомоторного развития, поведением, схожим с аутистическим, потерей навыков целенаправленного движения руками и эпилептическими припадками [1]. Частота встречаемости, как правило, составляет 1:15000 [3].
Впервые данное заболевание описывается в литературе второй половины ХХ века, когда австрийский педиатр Адресасс Ретт при исследовании группы девочек из разных городов Европы дал описание синдрому, который проявляется прогрессирующей потерей психических и двигательных навыков [1].
Из записок самого педиатра: «Это было в феврале 1965 года, когда я увидел двух матерей, держащих своих детей на коленях в приемной у врача. Оба ребенка раскачивались из стороны в сторону, а их мамы крепко держали их за руки. Я очень хорошо знал обеих девочек, которые в данный момент находились на обследовании. В это утро я, как обычно, проходил мимо них, и вдруг матери выпустили руки своих детей. Девочки сразу сцепили их вместе и, как будто по команде, стали совершать вдвоем одинаковые «моющие» движения. Это были одинаково прямые взгляды, одинаковые выражения лиц, одинаково слабая мускулатура и стереотипные движения. Я попросил женщин больше не ограничивать детей в движениях и был удивлен озарившей меня идеей…» [1]
Синдром Ретта возникает в результате нарушения развития головного мозга, причиной чего является мутация в гене транскрипции МЕСР2, связанного с Х хромосомой, находящейся в локусе Хq28 [2]. На сегодняшний день выявлено 8 мутаций данного гена. Предполагается, что ген МЕСР2 контролирует процессы развития ЦНС. Важно отметить, что мутации данного гена могут встречаться и у мальчиков, однако такие особи в популяции нежизнеспособны и умирают в первые часы жизни [1,2].
Согласно последним результатам нейрофизиологических исследований, при синдроме Ретта имеются нарушения со стороны синаптической организации, а также нарушения дофаминового обмена, обнаруживаются атрофические изменения в ростральных отделах коры головного мозга (КГМ) , которые возникают на ранних этапах онтогенеза [2]. Атрофические изменения также обнаруживаются в nucleus caudatus, substantia nigra. Нарушения в развитии КГМ проявляются грубыми нарушениями функций лобных долей мозга с выпадением их организующих влияний. Происходит деафферентация и растормаживание иерархически нижестоящих отделов коры и подкорковых структур, что ведет к регрессу психомоторного развития с возрастом [1,2].
Интервал проявления заболевания — от 6 до 20 лет, в крайне редких случаях первые симптомы выявляется в возрасте до 3 месяцев. Важным патогномоничным признаком является нормальное психомоторное развитие до начала заболевания [2]. Неспецифическими симптомами являются: врожденная гипотония, гипомимия, отставание в приобретении моторных навыков, слабое сосание, монотонный безутешный плач, эмоциональная ригидность, проблемы при фиксации взгляда [3].
Клинически синдром Ретта подразделяют на четыре стадии [2].
1. Первая стадия или стадия стагнации. Продолжительность от 2 до 4 месяцев, редко больше. В этой стадии происходит остановка психомоторного развития, замедление темпов роста головы, костей и стоп. Появляется мышечная гипотония, пропадает интерес к играм и окружающему миру, нарушается зрительное сосредоточение. На данной стадии клинический диагноз поставить невозможно [1].
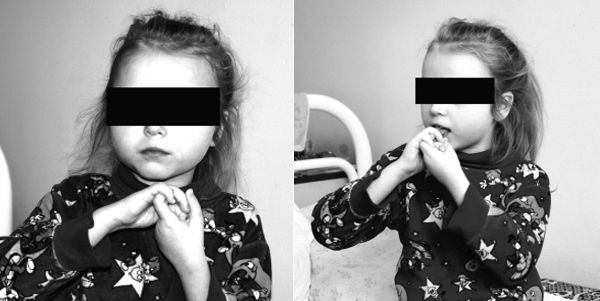
2. Вторая стадия или стадия быстрого регресса. В этот период происходит регресс психомоторного развития с постепенной утратой ранее приобретенных навыков. Ведущими симптомами являются потеря целенаправленной мануальной активности и появление особых стереотипных двигательных актов. Данный симптом является патогномоничным [1,2].
Под стереотипными двигательными актами подразумеваются: «моющие» движения руками, выкручивание кистей, трение рук друг об друга, а также постукивание зубами, оскал, жевательные движения. У детей возможно формирование лепетной речи или отдельные слова [3]. Однако во второй стадии заболевания происходит полная утрата экспрессивной речи, и дети способны издавать лишь отдельные звуки. Больные часто кричат, плохо засыпают. Подобная картина чаще всего формируется в возрасте до трех лет. Продолжительность жизни составляет несколько месяцев [1,2].
3. Третья стадия (псевдостационарная). Характеризуется относительной стагнацией развития заболевания. Происходит уменьшение симптомов аутистического поведения, проходят приступы беспокойства, нормализуется сон. В данной стадии клинически превалируют нарушения гнозиса и праксиса, сохраняются стереотипии в руках и отсутствие целенаправленной мануальной деятельности. Выявляются признаки нарушения проведения нервного импульса по пирамидному пути, статическая и динамическая атаксия, мышечная дистония, тремор [1,3].
Для данного этапа развития заболевания характерны 3 разновидности пароксизмальных состояний: эпизоды гипервентиляции, синкопальные состояния и эпилептические приступы [3].
4. Четвертая стадия или стадия поздних двигательных нарушений. Данная стадия характеризуется прогрессированием развития моторных расстройств и, как правило, проявляется ближе к 10 годам. Усугубляются парезы, нарастает выраженность спастико-амиотрофического синдрома. Хорошо заметна мышечная ригидность в нижних конечностях. Увеличивается степень сколиоза, кифосколиоза. К 15 годам ребенок не способен самостоятельно ходить [1].
Основным методом объективного исследования данного заболевания является ЭЭГ. На нем характерно замедление основной активности фоновой записи [1]. При этом признаки эпилепсии появляются на ЭЭГ в виде комплексов «пик-волна», «острая» волна, медленных комплексов «острая-медленная» волна задолго до самих приступов. На МРТ отмечают уменьшение объема мозга преимущественно за счет уменьшения белого вещества, атрофические изменения хвостатого ядра [1].
Таблица 1 | Диагностические критерии типичного синдрома Ретта [1]
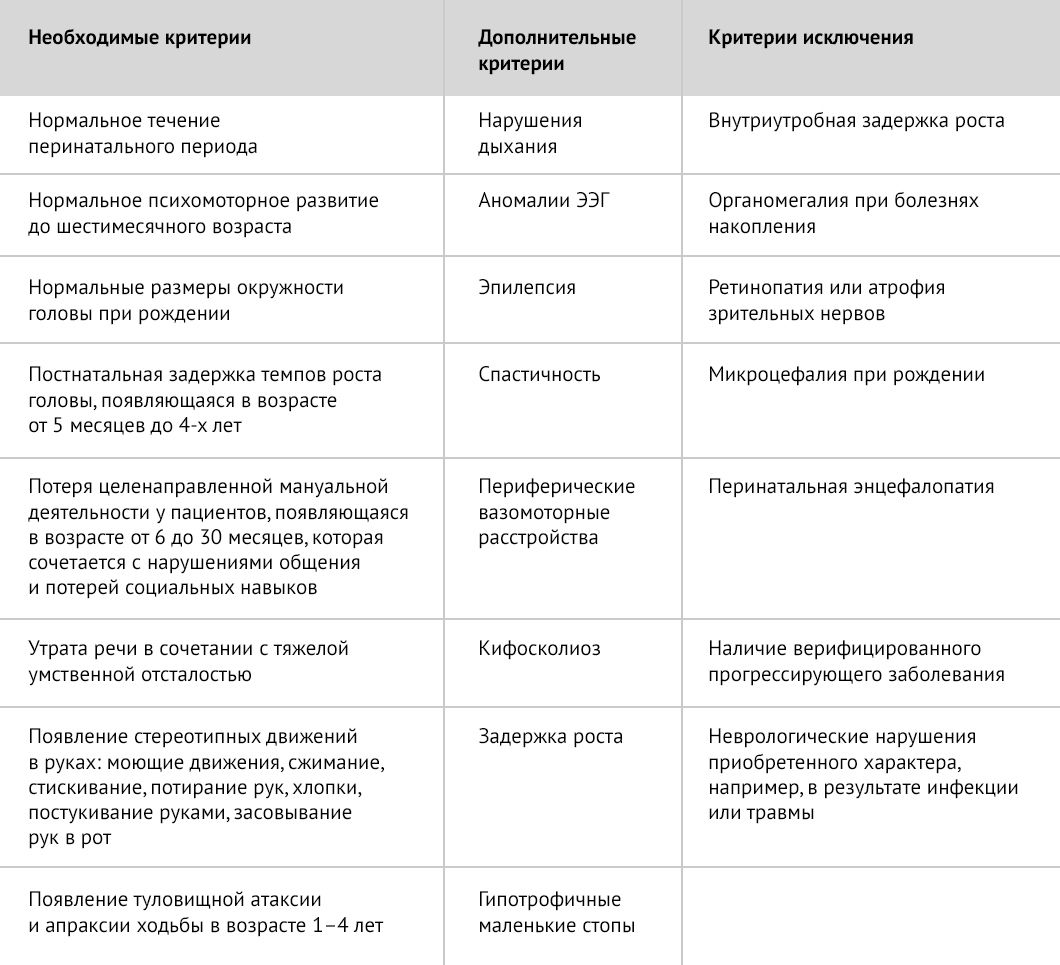
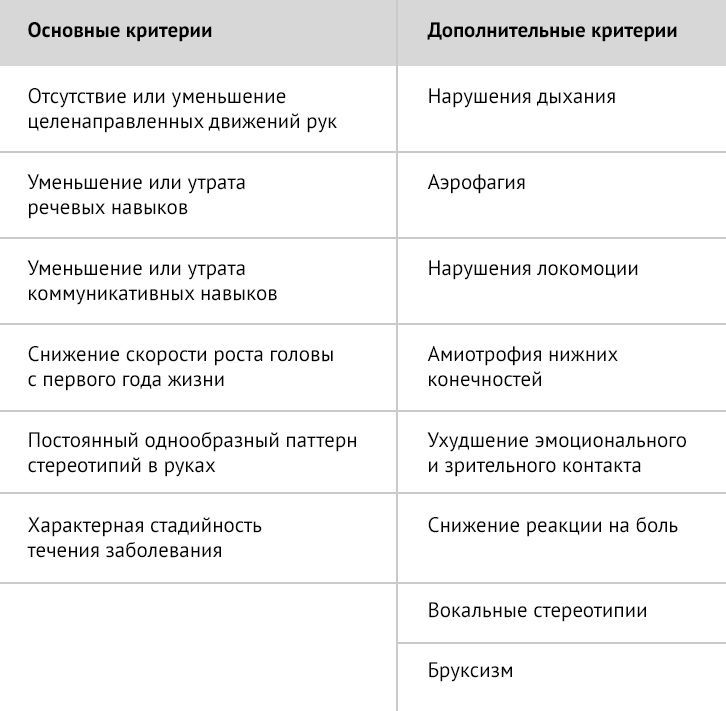
Таблица 2 | Диагностические критерии атипичного синдрома Ретта [1]
Главной задачей в лечении синдрома Ретта является контроль эпилептических припадков. Лучшие препараты для этих целей — карбамазепин, ламотриджин, топирамат [1].
.
Источники:
- К.Ю. Мухин. Синдром Ретта / К.Ю. Мухин, В.И. Карпова, И.С. Безрукова // Российский журнал детской неврологии — 2015 — № 10 — С. 43–51
- И.Ю. Юров. Комплексный клинико-генетический подход к диагностике синдрома Ретта у детей / И.Ю. Юров, С.Г. Ворсанова, В.Ю. Воинова-Улас, П.В. Новиков, Ю.Б. Юров // Российский журнал педиатрии — 2012 — №6 — С. 38–43
- О.В. Тимуца. Синдрома Ретта: социально-психологический аспект проблем людей с редкими заболеваниями / О.В. Тимуца, В.Д. Менделевич // Неврологический вестник — 2013 - №2 — С.60-65
