
Тубулопатии, сопровождающиеся потерей солей
Тубулопатии со вторичным гиперальдостеронизмом, сопровождающиеся потерей солей (ТСПС), обусловлены четко определенными наследственными патологиями канальцев почки. В патогенезе ТСПС в основном участвуют два сегмента дистального отдела нефрона: восходящее колено петли Генле (ВКПГ) и дистальный извитой каналец (ДИК). Функции сегментов до и после плотного пятна весьма различны, и это оказывает существенное влияние на клиническую картину патологий петли Генле и ДИКа — синдромов, подобных Барттеру и Гительману. Дефекты водонепроницаемой части восходящего колена, главная функция которого — реабсорбция солей, приводят к значительным потерям электролитов и воды, аналогично эффектам петлевых диуретиков (фуросемид). Напротив, дефекты в ДИКе, с его незначительной способностью к реабсорбции соли, учитывая его главную функцию (контроль экскреции кальция и магния), вызывают хронический дисбаланс электролитов, аналогичный последствиям хронического лечения тиазидами.
Наиболее тяжелое состояние развивается при сочетании патологий петли Генле и ДИКа. Оно сходно с усиленным мочегонным эффектом от совместного лечения петлевыми диуретиками и тиазидами. Помимо использования солевых растворов в терапии пациентов с данной патологией, ингибиторы простагландин E2-синтазы (например, чистый фуросемид) являются наиболее эффективным терапевтическим вариантом при полиурических расстройствах петли Генле, особенно у недоношенных детей с обезвоживанием и снижением ОЦК. При патологиях ДИКа блокаторы ренин-ангиотензин-альдостероновой системы (РААС), которые могут рекомендоваться после приема соли, калия и магния, считаются недостаточными. Похоже, что большинству пациентов с ТСПС на протяжении всей жизни требуется комбинация различных препаратов.
Наследственные тубулопатии: синдром Барттера и Гительмана
Синдромы Барттера и Гительмана являются редкими наследственными тубулопатиями, сопровождающимися потерей солей (ТСПС), что приводит к гипокалиемии. Они характеризуются дисфункцией трансэпителиального транспорта электролитов в восходящем колене петли Генле или дистальном извитом канальце (патологии ДИКа). Также встречается сочетание этих патологических процессов (комбинированные нарушения). Гипокалиемический алкалоз служит отличительной чертой патологий петли Генле и ДИКа и позволяет отличить их от тубулопатий, сопровождающихся потерей соли, в других отделах нефрона.
Синдром Гительмана является гораздо более распространенным заболеванием, чем синдром Барттера. В докладе из исследования Framingham Heart Study распространенность для синдрома Гительмана составляла 1:40 000 случаев по сравнению с 1:1 000 000 случаев для синдрома Барттера. Более низкая распространенность синдрома Барттера в популяции может быть частично обусловлена пренатальной или неонатальной смертностью, вызванной данной патологией, вследствие которой синдром не был диагностирован.
Общий обзор
Функции ВКПГ и ДИКа различны, а значит, патология этих сегментов нефрона вызывает совершенно разные изменения. Для оценки их отличий необходимо понимать основные механизмы транспорта электролитов и воды через эпителиальные клетки в трех основных частях дистальных канальцев: восходящее колено петли Генле, ДИК и дистальный отдел нефрона, чувствительный к альдостерону (ДОНЧА), включающий конечную часть ДИКа, трубочки, соединяющие ДИК и собирательные трубочки, и собирательные трубочки.
В восходящем колене петли Генле ключевым звеном в активном транспорте ионов Na+ и Cl– в клетки почечного канальца является чувствительный к фуросемиду натрий-калий-2-хлорид-котранспортер NKCC2. Ионы Na+ активно откачиваются из клеток восходящего колена петли Генле посредством базолатеральной Na+/K+-ATФазы, в то время как как Cl– покидает базальный полюс клетки через определенные каналы-переносчики ионов хлора (ClC-Ka и ClC-Kb). Для правильной работы обоих каналов-переносчиков ионов хлора требуется b-субъединица барттина. В отличие от ионов Na+ и Cl–, K+ повторно возвращается обратно в жидкость канальца через апикальную мембрану с помощью проницаемого для калия ионного канала — почечного наружного медуллярного калиевого канала (ПНМКК). Таким образом, неповрежденный ПНМКК необходим для создания положительного трансэпителиального потенциала просвета, который используется для активной реабсорбции трансцеллюлярной соли, а также для поддержания движущей силы пассивной парацеллюлярной транспортировки ионов Ca2+ и Mg2+ в восходящее колено петли Генле.
ДИК, который начинается после плотного пятна, играет важную роль в почечной экскреции хлорида натрия (около 10 % отфильтрованной нагрузки), а также кальция и магния (приблизительно по 8–10 % каждого). Как и в восходящем колене петли Генле, активный транцеллюлярный перенос соли в ДИКе обеспечивается за счет деятельности базолатеральной Na+/K+-АТФазы. В основе активного транспорта ионов Na+ в клетки в начале ДИКа лежит работа апикально экпрессированного тиазид-чувствительного натрий-хлор-котранспортера NCCT. В этой части ДИКа активность Na+/K+-АТФазы дополняется калиевым каналом внутреннего выпрямления Kir4.1, который облегчает базолатеральный выход ионов Na+. После внутриклеточного поглощения вместе с ионами Na+ ионы Cl– покидают клетку через ClC-Kb (ClC-Ka не экспрессирован в ДИКе). Как следствие нарушения транспорта ионов Na+, при любой патологии ДИКа внутриклеточная концентрация Na+ может быть понижена, что должно повысить поглощение ионов Na+ и увеличить экскрецию ионов Са2+ через клеточную мембрану в почечный интерстиций. Независимо от патогенеза гипокальциурия является отличительной чертой патологии ДИКа.
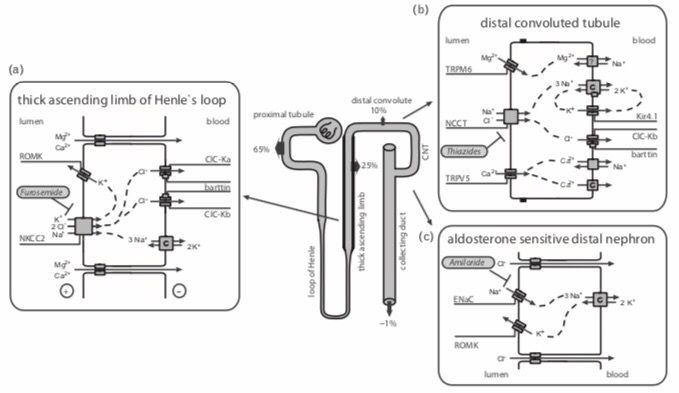 Рисунок 1 ❘ Реабсорбция растворенных веществ в восходящем колене петли Генле (ВКПГ), дистальном извитом канальце (ДИКе) и дистальном отделе нефрона, чувствительному к альдостерону (ДОНЧА), включая позднюю часть ДИКа, трубочки, соединяющие ДИК и собирательные трубочки, и собирательные трубочки.
Рисунок 1 ❘ Реабсорбция растворенных веществ в восходящем колене петли Генле (ВКПГ), дистальном извитом канальце (ДИКе) и дистальном отделе нефрона, чувствительному к альдостерону (ДОНЧА), включая позднюю часть ДИКа, трубочки, соединяющие ДИК и собирательные трубочки, и собирательные трубочки.
В ВКПГ(а) NaCl реабсорбируется фуросемид-чувствительным NKCC2 каналом вместе с калием, который затем рециркулирует через ROMK калиевые каналы. Кальций и магний пассивно реабсорбируются через парацеллюлярный путь.
В ДИКе (b) реабсорбция соли происходит через котранспортер NCCT. Как и в ВКПГ, Na+ реабсорбируется в базолатеральном полюсе клетки с помощью Na+/K+-АТФазы, и Cl– покидает клетку через хлорные каналы. В этой части нефрона работа Na+/K+-АТФазы дополняется калиевыми каналами Kir4.1, которые облегчают базолатеральную реабсорбцию Na+ путем рециркуляции калия обратно в почечный интерстиций. Реабсорбция магния и кальция в ДИК является активной и обусловлена транспортом через селективные ионные каналы (TRPM6 и TRPV5 соответственно). В ДОНЧА калиевые каналы ROMK, в дополнение к их роли в ВКПГ, необходимы для секреции ионов калия в обмен на реабсорбцию натрия через эпителиальные натриевые каналы (ENaC) под влиянием альдостерона.
Классификация
Согласно патофизиологическиму и фармакологическому подходам, ТСПС могут быть разделены на две основные группы патологий канальцев почек: дефекты водонепроницаемой части восходящего колена, аналогичные эффекту петлевых диуретиков (фуросемид), и дефекты в ДИКе, аналогичные последствиям хронического лечения тиазидами. Первая группа далее подразделяется на патологии ВКПГ, вызванные генетическим дефектом в гене NKCC2 или гене ROMK (L1 и L2 соответственно). Вторая группа включает в себя нарушения, связанные с генетической патологией в каналах NCCT, ClC-Kb или Kir 4.1 (DC1, DC2 или DC3 соответственно). Генетические патологии ВКПГ также называются синдромом Барттера типа I и типа II, а патологии ДИКа — синдромом Гительмана, синдромом Барттера тип III и синдромом EAST. Тип L1 представляет собой фуросемид-подобную ТСПС, тогда как тип L2 представляет собой, прежде всего, фуросемид-подобную ТСПС с транзиторной гиперкалиемией из-за недостатка экспрессии ROMK канала в ДИКе, трубочках, соединяющих ДИК и собирательные трубочки, и собирательных трубочках, где это необходимо для секреции калия.
Комбинация патологий ДИКа и ВКПГ является одной из самых тяжелых категорий ТСПС, и это состояние образует третью группу: фуросемид-тиазидоподобную ТСПС (тип L-DC). Генетически это комбинированное расстройство относится к синдрому Барттера с сенсоневральной глухотой. В настоящее время эта группа патологий подразделяется далее на L-DC1 (синдром, вызванный комбинированными генетическими дефектами в каналах ClC-Ka и ClC-Kb) и L-DC2 (синдром, вызванный генетическим дефектом в b-субъединице барттина).
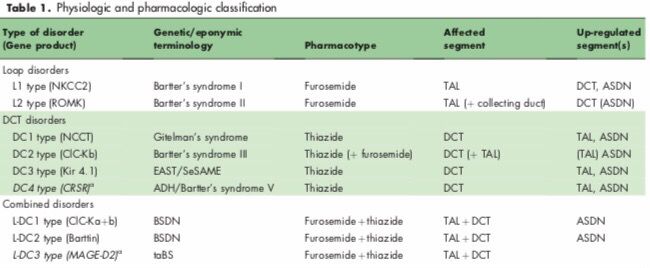 Рисунок 2 ❘ Сегмент в круглых скобках имеет второстепенное значение для клинического проявления.
Рисунок 2 ❘ Сегмент в круглых скобках имеет второстепенное значение для клинического проявления.
ADH — аутосомно-доминантная гипокальциемия; ASDN — дистальный отдел нефрона, чувствительный к альдостерону; BSDN — синдром Барттера с сенсоневральной глухотой; CaSR — кальциевый рецептор; DCT — ДИК; EAST/SeSAME — синдром, который сопровождает эпилепсия, атаксия, сенсорная глухота и тубулопатия; MAGE-D2 — меланома-ассоциированный антиген D2; taBS — переходный пренатальный синдром Барттера; TAL — ВКПГ.
Генетика и клинические проявления
Различные варианты синдрома Барттера — редкие наследственные нарушения, которые охватывают гетерогенную группу ТСПС, поражающих ВКПГ, с разнообразными причинами, но с общей точкой патогенеза — потерей функции NKCC2 канала. Среди фенотипов синдрома Барттера наиболее распространены мутации, которые влияют на ген KCNJ1 (канал ROMK), реже — на ген SLC12A1 (канал NKCC2), ген CLCNKB (канал ClC-Kb) и ген BSND (белок барттин). Нефрокальциноз типичен для фенотипов с мутациями генов KCNJ1 и SLC12A1. Хроническая почечная недостаточность, хоть встречается и редко, но все же обнаруживается среди фенотипов с мутациями в генах KCNJ1, CLCNKB и BSND (при этом нефрокальциноз встречается не всегда). Потеря слуха происходит только у пациентов с мутациями гена BSND.
Синдром Гительмана, также известный как семейная гипокалиемия с гипомагниемией, является одной из наиболее частых наследственных тубулопатий и характеризуется гипокалиемическим метаболическим алкалозом со значительной гипомагниемией и гипокальциурией. Болезнь вызвана мутациями гена SLC12A3, расположенного в локусе 16q13 16-й хромосомы, и имеет аутосомно-рецессивный тип наследования. Ген SLC12A3 кодирует NCC белок, экспрессирующийся в начальной части ДИКа, функция которого подавляется тиазидными диуретиками.
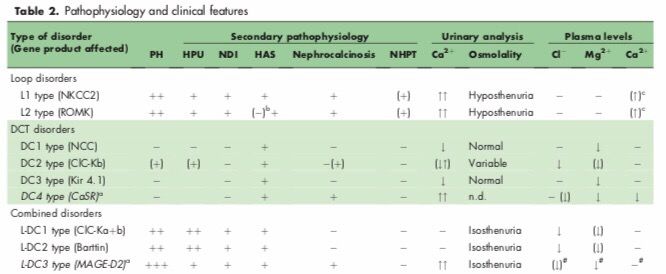 Рисунок 3 ❘ При всех перечисленных заболеваниях присутствует гипокалиемия и алкалоз. Стрелки вверх и вниз указывают соответственно на увеличение или уменьшение экскреции или концентрации. Знаки в круглых скобках указывают на то, что либо данный патофизиологический признак является вторичным и протекает в легкой форме, и/или изменения редко присутствуют, и/или незначительно отличаются от нормальных показателей.
Рисунок 3 ❘ При всех перечисленных заболеваниях присутствует гипокалиемия и алкалоз. Стрелки вверх и вниз указывают соответственно на увеличение или уменьшение экскреции или концентрации. Знаки в круглых скобках указывают на то, что либо данный патофизиологический признак является вторичным и протекает в легкой форме, и/или изменения редко присутствуют, и/или незначительно отличаются от нормальных показателей.
n. d. — не определено. CaSR — кальциевый рецептор; ClC — хлорный канал; HAS — гиперальдостеронизм; HPU — гиперпростагландинурия; MAGE-D2 — меланома-ассоциированный антиген D2; NCC — хлорангидрид натрия; NDI — нефрогенный несахарный диабет; NHPT — гиперпаратиреоз новорожденных; NKCC2 — натрий-калий-2-хлорид-котранспортер; PH — полигидрамнион; РОМК — внешний медуллярный калиевый канал.
Терапия
Общим в терапии как патологий ВКПГ, так и патологий ДИКа является восполнение потерянной жидкости (насколько это возможно). Цель должна состоять в том, чтобы восстановить баланс электролитов, пул минеральных веществ в организме и объем внеклеточной жидкости. Это самый эффективный способ лечения метаболического алкалоза. В остальном подход к лечению патологий ДИКа и ВКПГ значительно отличается. У младенцев с жизнеугрожающими патологиями ВКПГ восстановление баланса воды и электролитов (предпочтительно через центральный венозный катетер) является первой терапевтической мерой для предотвращения шока и острой почечной недостаточности. Однако необходимо понимать, что чрезмерная гемодилюция усугубит полиурию. Поскольку канальцевый ток мочи связан, по крайней мере частично, со стимуляцией синтеза почечного PGE2, показана антидиуретическая терапия с ингибитором синтеза PGE2 (например, индометацин). Из-за растущей уязвимости слизистой оболочки желудка к неселективным нестероидным противовоспалительным средствам (НПВС) селективные ингибиторы циклооксигеназы-2, например, целекоксиб, можно рассматривать в качестве терапии для детей и молодых взрослых с патологиями желудочно-кишечного тракта. У детей старшего возраста и взрослых пациентов с медленно развивающейся патологией ДИКа диета, богатая солью и калием, является основой краткосрочного и долгосрочного лечения. Однако следует отметить, что терапия с использованием магния предпочтительна в виде модифицированных органических солевых препаратов; она рекомендуется для эффективного лечения и профилактики осложняющей основное заболевание гипокалиемии. В частности, у пациентов с патологиями ДИКа и хондрокальцинозом, очевидно, связанным с гипомагниемией, отмечены улучшения в состоянии при назначении магния. Кроме того, после назначения магния значительно улучшается состояние беременных женщин, больных синдромом Гительмана, особенно с гиперемезисом (неукротимой рвотой).
Источники
1. Seyberth H. W., Weber S., Kömhoff M. Bartter's and Gitelman's syndrome //Current opinion in pediatrics. – 2017. – Т. 29. – №. 2. – С. 179-186.
2. Naesens M. et al. Bartter’s and Gitelman’s syndromes: from gene to clinic //Nephron Physiology. – 2004. – Т. 96. – №. 3. – С. p65-p78.
3. Koulouridis E., Koulouridis I. Molecular pathophysiology of Bartter’s and Gitelman’s syndromes //World Journal of Pediatrics. – 2015. – Т. 11. – №. 2. – С. 113-125.
