
Синдром Чедиака-Хигаси

Синдром Чедиака-Хигаси — редкое аутосомно-рецессивное заболевание, которое характеризуется тяжелым врожденным иммунодефицитом по причине дефекта гена CHS1/LYST. Проявляется болезнь частыми бактериальными инфекциями, коагулопатией, нарушениями пигментации кожи, а также прогрессирующими неврологическими расстройствами [1].
Иммунологические нарушения выражаются в нейтропении и нехватке естественных киллеров (NK-клеток). Зачастую заболевание оказывается летальным — большая часть пациентов умирает в детстве. Заподозрить синдром Чедиака-Хигаси можно по тем же признакам, что и любой врожденный иммунодефицит, однако имеется и довольно специфичный критерий — гигантские внутриклеточные включения (или органеллы) в популяциях разных клеток и определенные дефекты в них [2].
Чтобы более наглядно описать происходящие процессы при данном заболевании, еще во второй половине прошлого столетия была предложена оригинальная модель «бежевой мыши». Вследствие мутации в определенном гене (в то время конкретная точка генома, разумеется, не была известна) пигментация экспериментальной мыши изменялась, и исследователи определили ее цвет как «бежевый» («beige»). Помимо цвета у таких животных имелся еще ряд аномалий в виде огромных гранул меланина в лимфоцитах, нейтрофилах и эозинофилах (рис. 1), также подобные включения наблюдались в ряде других клеток [3].
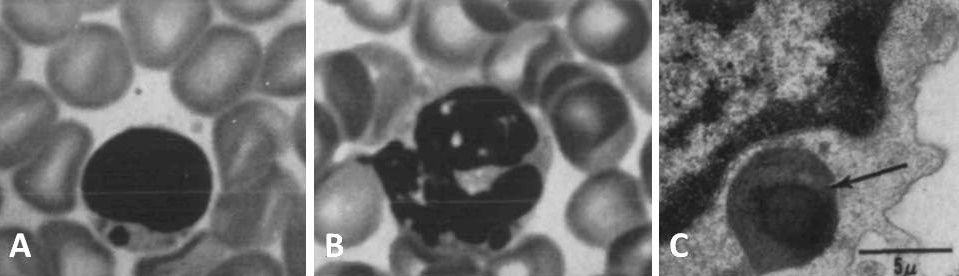
Рисунок 1 | Периферическая кровь бежевой мыши. Световая микроскопия: определяются гранулы в цитоплазме лимфоцита (А) и нейтрофила (В). Электронная микроскопия: объемная гранула с темным содержимым в цитоплазме лимфоцита. Относится к непостоянным аномальным включениям [3].
Позднее данный ген у мыши был идентифицирован и назван Lyst (кодирует регулятор лизосомального транспорта — lysosomal trafficking regulator), человеческий ген обозначают как LYST. Фермент, за экспрессию которого отвечает LYST, обеспечивает нормальный экзо- и эндоцитоз лизосомы; в случае мутации лизосома не может выполнять свои функции [4].
Сегодня этот ген настолько прочно ассоциируется с синдромом Чедиака-Хигаси, что в современной литературе его обозначают как CHS1/LYST(человеческий ген Chediak-Higashi syndrome-1 или LYST — оба варианта равнозначны) [1].
Роль регулятора лизосомального транспорта (LYST)
Данный белок кодируется достаточно объемным геном LYST, который содержит 55 экзонов и расположен в локусе 1q43. Большая длина нуклеотидной последовательности этого гена — фактор неоднозначный: с одной стороны, это существенно затрудняет диагностику, с другой — большое количество мутаций вообще никак не влияет на жизнедеятельность человека и не вызывает синдром Чедиака-Хигаси [5].
Ген кодирует одноименный большой каталитический белок — регулятор лизосомального транспорта (425 kDa, 3801 аминокислотное основание). Протеин относится к семейству BEACH — (Beige and Chediak-Higashi) — и, по-видимому, стал первым его представителем [4]. Белки данного семейства имеют три общих С-концевых домена: PH (Pleckstrin-homology домен), BEACH (Beige and Chediak-Higashi домен) и WD40-повторения (рис. 2) [5].
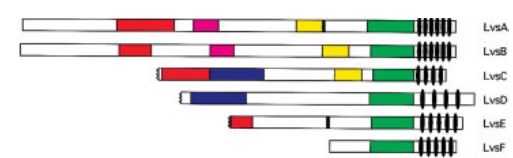
Рисунок 2 | Строение шести родственных белков семейства BEACH слизевика рода Dictyostelium [6].
На рис. 2 изображена диаграмма, наглядно показывающая общность строения шести белков семейства BEACH (в данном случае — шесть белков слизевика рода Dictyostelium). Зеленым цветом обозначен собственно BEACH-домен, наиболее консервативный во всех белках. «Овалами» у С-конца обозначены WD40-повторения — участки, предположительно вовлеченные в белок-белковые взаимодействия. Другими цветами показаны иные идентичные аминокислотные последовательности [6].
Сегодня известно, что белки данного семейства есть у разных представителей животного мира. Имея примерно одинаковое строение, они выполняют многочисленные и крайне сложные функции, обсуждение которых выходит за рамки данной темы.
Собственно LYST обеспечивает секрецию лизосомальных ферментов. Без белка-регулятора транспорт протеолитических ферментов невозможен, вследствие этого цитотоксическая активность фагоцитарных клеток нарушается. Этим также объясняется нарушение транспорта различных метаболитов (накопление в клетках гранул с меланином, что обеспечивает яркий клинический признак синдрома Чедиака-Хигаси), а также нарушения метаболизма некоторых других белков, что обусловливает клиническую картину заболевания и сопутствующие патологии, точный механизм развития которых все еще неизвестен [1,3].
Стоит также заметить, что в последнее время появляются сообщения о преимущественном влиянии дефекта LYST именно на NK-клетки. По последним данным, в лимфоцитах происходит нарушение экзоцитоза протеолитических ферментов, хотя цитокиновая регуляция остается в норме [7].
Клиническая картина
Существует два основных клинических проявления синдрома Чедиака-Хигаси: «парциальный альбинизм» и рецидивирующие пиогенные инфекции. Оба, на первый взгляд, не связанных между собой критерия имеют единую причину — дефект LYST, вследствие чего нарушается транспорт меланина и лизосомальных ферментов.
Цвет волос пациента может быть от сероватого до буквально седого — зависит от этнической принадлежности пациента. Также нарушается пигментация радужной оболочки, следствием чего становится фоточувствительность и сниженная функция зрительного аппарата.
Рецидивирующие бактериальные (в том числе оппортунистические) инфекции часто поражают дыхательные пути, кожу. Возможны дефекты свертывающей системы из-за нарушенного синтеза тромбоцитов, проявляется это небольшими кровоподтеками (зачастую — на слизистых), однако иногда развивается полноценный геморрагический синдром.
Помимо этого возможны неврологические нарушения: атаксия, сенсорные расстройства, прогрессирующая нейродегенерация [1].
Первые проявления заболевания начинаются с раннего детства. Наиболее опасными осложнениями считаются инфекции, для предупреждения которых допустима профилактическая антибиотикотерапия. И все же большинство пациентов умирает в первые десять лет жизни. Наиболее распространенными причинами смерти являются кровотечения, инфекции или гемофагоцитарный лимфогистиоцитоз [7].
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз (ГФЛГ) — патологическое состояние, которое может быть вызвано разнообразными причинами, часто — первичными иммунодефицитами (к числу которых относится и синдром Чедиака-Хигаси). Суть данного состояния — гиперпродукция гистиоцитов и иммунокомпетентных клеток [5].
Как уже было сказано выше, мутация LYST нарушает цитотоксическую функцию клеток, но не метаболизм регуляторных факторов. В ответ на антиген развивается обыкновенная воспалительная реакция — однако элиминация чужеродного агента невозможна. В связи с этим продолжается продукция провоспалительных факторов (ИФН-γ, ФНО, интерлейкины), что увеличивает количество лимфоцитов, нейтрофилов, повышает активность макрофагов. Гиперпродукция цитокинов, не ингибированная по механизму обратной связи, иногда называется «цитокиновым штормом» [5].
Следствием этого является лимфогистиоцитарная инфильтрация различных тканей с развитием в них разнообразных повреждений, а макрофаги могут разрушать нормальные функционирующие клетки (в том числе — форменные элементы крови) [5].
Диагностика синдрома Чедиака-Хигаси и сопутствующих заболеваний
В диагностировании синдрома Чедиака-Хигаси особых сложностей не возникает. Как уже было сказано выше, основная задача клинициста — распознать врожденное иммунодефицитное состояние, дальнейшая диагностика основана на данных иммунограммы и генетического анализа. Достаточно специфичным признаком является накопление больших внутриклеточных везикул в различных клетках, в том числе — лейкоцитах [2].
Одним из наиболее серьезных осложнений при данном синдроме является развитие ГФЛГ. Диагностические критерии при этом патологическом состоянии можно представить следующим образом [5]:
- Наличие генетического дефекта, связанного с ГФЛГ (зачастую это первичные иммунодефициты).
- Наличие как минимум пяти из нижеперечисленных критериев:
- лихорадка;
- спленомегалия;
- цитопения хотя бы в двух клеточных популяциях:
а) гемоглобин < 90 г/л (для новорожденных — < 100 г/л);
б) тромбоциты < 100×109/л;
в) нейтрофилы < 1×109/л;
- гипертриглицеридемия (> 3 ммоль/л) или гипофибриногенемия (< 1,5 г/л);
- гиперферритинемия > 500 мкг/л;
- растворимые молекулы CD25 > 2400 Ед/мл;
- гемофагоцитоз в костном мозге, селезенке, лимфоузлах
- низкая (вплоть до полного отсутствия) цитотоксическая активность NK-клеток.
Важно отметить: ГФЛГ является настолько частым серьезным осложнением, что в современной литературе принято разделять синдром Чедиака-Хигаси на две формы: «классическую» (с развитием ГФЛГ) и «атипичную» (без такового) [5].
Лечение синдрома Чедиака-Хигаси и сопутствующих патологий
Как и в большинстве наследственных иммунодефицитов, вариантов терапии немного. Наиболее распространенным методом лечения врожденных иммунологических нарушений является пересадка гемопоэтических клеток; данный синдром не является исключением.
В 2007 году в «Bone Marrow Transplantation» появилась публикация, авторы которой сообщали о 35 случаях проведения пересадки гемопоэтических клеток пациентам с синдромом Чедиака-Хигаси [8]. Перед проведением операции лечение осуществлялось в лучшем случае патогенетическое, призванное замедлить прогрессирование заболевания, избежать осложнений и подготовить пациента к пересадке гемопоэтических клеток.
Всего 13 пациентов получили материал от HLA-идентичного донора (родного брата или сестры), 10 — от родственника, еще 12 — от несвязанного донора. По результатам проведенной терапии, 27 (77 %) из 35 пациентов достигли ремиссии, однако 5-летняя выживаемость составила 62 % (22 пациента).
Наиболее распространенными причинами смерти были посттрансплантационные осложнения и хронические заболевания. Среди умерших — большинство получило аллотрансплантат от не полностью подходящего донора.
Таким образом, исследователи пришли к выводу, что подобная методика может быть достаточно эффективной, если донор и реципиент являются родственниками. Также следует отметить, что шесть пациентов на момент проведения операции были старше девяти лет, что свидетельствует о существенных успехах в сдерживании и предотвращении развития осложнений у пациентов (одному из реципиентов на момент трансплантации было 19 лет).
На современном этапе пересадка гемопоэтических клеток является наиболее эффективным способом лечения синдрома Чедиака-Хигаси [9]. Вероятно, в будущем станет возможна коррекция непосредственно LYST с помощью генно-инженерных методик (например, подобная терапия уже существует для хронической гранулематозной болезни — другого наследственного иммунодефицитного состояния [10]), однако пока что такой альтернативы для синдрома Чедиака-Хигаси нет.
Часто синдром Чедиака-Хигаси осложняется ГФЛГ — тяжелым состоянием, которое требует немедленного лечения. Среди наиболее эффективных методов терапии в гайдлайне 2004 года описаны химиотерапия и пересадка гемопоэтических клеток [11].
Также на протяжении лечения, до и после него в профилактических целях используется антибиотикотерапия [1,11].
Источники:
- J. Kaplan, I. De Domenico, and D. M. Ward, ‘Chediak-Higashi syndrome’, Curr. Opin. Hematol., vol. 15, pp. 22–29, 2008.
- D. L. Nagle et al., ‘Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome’, Nat. Genet., vol. 14, no. 3, pp. 307–311, 1996.
- M. A. Lutzner, C. T. Lowrie, and H. W. Jordan, ‘Giant granules in leukocytes of the beige mouse’, J. Hered., vol. 58, no. 6, pp. 299–300, 1967.
- M. D. F. S. Barbosa et al., ‘Identification of the homologus beige and Chediak-Higashi syndrome genes’, Nature, vol. 382, pp. 262–265, 1996.
- S. Ehl and G. de Saint Basile, ‘Genetic Diseases Predisposing to HLH’, Stiehm’s Immune Defic., pp. 437–460, 2014.
- N. Wang, W. I. Wu, and A. De Lozanne, ‘BEACH family of proteins: Phylogenetic and functional analysis of six Dictyostelium BEACH proteins’, J. Cell. Biochem., vol. 86, no. 3, pp. 561–570, 2002.
- A. Gil-Krzewska et al., ‘Chediak-Higashi syndrome: Lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells’, J. Allergy Clin. Immunol., vol. 137, no. 4, pp. 1165–1177, 2016.
- M. Eapen et al., ‘Hematopoietic cell transplantation for Chediak-Higashi syndrome’, Bone Marrow Transplant., vol. 39, no. 7, pp. 411–415, 2007.
- M. L. Lozano, J. Rivera, I. Sánchez-Guiu, and V. Vicente, ‘Towards the targeted management of Chediak-Higashi syndrome’, Orphanet J. Rare Dis., vol. 9, no. 1, 2014.
- M. G. Ott et al., ‘Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1’, Nat. Med., vol. 12, no. 4, pp. 401–409, 2006.
- Jan-Inge Henter, ‘HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocutic Lymphohistiocytosis’, Pediatr. Blood Cancer, vol. 48, pp. 124–131, 2007.
