
PTEN как метаболический регулятор. Часть 2: опухолевая ткань

Многообразие функций и неоднозначность взаимосвязи сигналов клеточных молекулярных путей формируют сложную динамическую систему. Попытки упростить понимание роли конкретной молекулы в этих каскадах неизбежно приводят к ограничению ее значимости, а с этим и к использованию не всего потенциала воздействия на этот компонент. Максимальный эффект от модулирования клеточной активности возможен лишь при осознании комплексности каждого вмешательства и широком множестве последствий.
Длительным заложником одной роли был ген и его белковый продукт PTEN (гомолог тензина и фосфатазы). Изначально он определен как классический онкосупрессор. Его функциональная активность заключается в подавлении сигналов молекулярного пути Akt, ответственного не только за пролиферацию и выживание клеток, но и, как обнаружилось позднее, также вовлеченного в системный гомеостаз. PTEN представляет собой двойную (протеиновую и липидную) фосфатазу, основным субстратом которой является фосфатидилинозитол-3,4,5-трифосфат (PIP3), продукт PI3-киназы. Снижение уровня PIP3 под действием PTEN не позволяет рекрутировать серин-треониновую протенкиназу Akt на поверхность клеточной мембраны и не допускает ее активации. Реализующиеся в итоге блокирования Аkt-пути метаболические эффекты, такие как повышение количества белков-переносчиков глюкозы на клеточной мембране, активация гексокиназы и фосфофруктокиназы, а также индукция липогенеза de novo параллельно с β-окислением жирных кислот, способствуют канцерогенезу [1].
Состояние инсулинорезистентности подразумевает снижение подавляющего действия инсулина на глюконеогенез в печени. Постоянно высокий уровень синтеза глюкозы приводит к постпрандиальной гипергликемии. В то же время гиперинсулинемия в печени вызывает липогенез, что приводит к накоплению гранул липидов в гепатоцитах (стеатоз). Этот феномен является одним из морфологических признаков синдрома инсулинорезистентности и отображает так называемую «селективную печеночную инсулинорезистентность».
Утрата экспрессии PTEN в печени приводит к формированию неалкогольного стеатогепатита (НАСГ) по другим механизмам на фоне подавления глюконеогенеза. Усиление активности пути PI3K/Akt у таких организмов приводит к улучшению способности печени метаболизировать глюкозу, превращая печень в «приемник глюкозы». Это повышает способность организма справляться с гликемическим стрессом. Но также потеря PTEN активирует синтазу жирных кислот и, соответственно, усиливает липогенез de novo [2]. Часть вновь синтезированных жирных кислот вовлекается в процесс β-окисления. Значительное число внутриклеточных метаболитов, вовлеченных в синтез жирных кислот de novo и их β-деградацию, обнаружены в клеточных линиях рака предстательной железы [3].
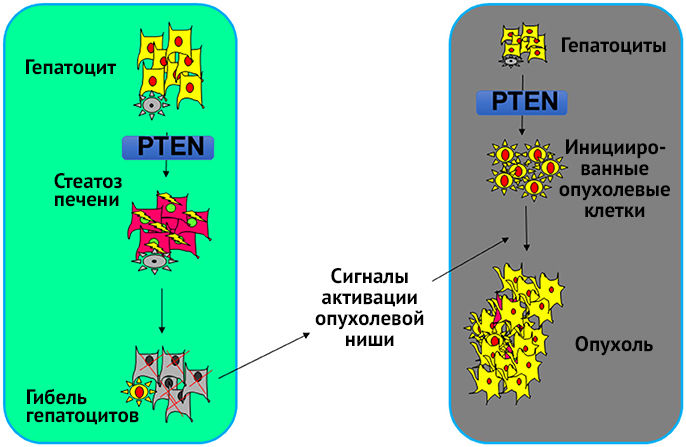
НАСГ создает благоприятные условия для повреждения паренхимы печени. Обнаружено, что ингибирование НАСГ с помощью диеты или генетической делеции метаболически активного подтипа Akt (Akt2) тормозит развитие опухоли. Таким образом, печеночный стеатоз формирует благоприятную для опухоли среду (рис.1) [4]. Значительную роль в канцерогенезе играет воспаление, которое неизбежно присоединяется к стеатозу (стеатоз + воспаление = стеатогепатит). Сигнал Wnt, продуцируемый макрофагами в клеточном воспалительном инфильтрате, является одним из важнейших нишевых сигналов формирующейся опухоли [5].
В отличие от хорошо дифференцированных клеток, которые в основном зависят от энергии митохондриального окислительного фосфорилирования, опухолевые клетки предпочитают аэробный гликолиз. Они генерируют энергию путем преобразования глюкозы в лактат в аэробной среде. Это парадоксальное явление называется эффектом Варбурга [6].
При дефектах PTEN перепрограммирование на этот гликолитический путь происходит за счет ряда механизмов. Во-первых, происходит активация синтеза гексокиназы II (HK II), которая катализирует первую стадию гликолитического пути. Также эффект Варбурга стимулирует фруктозо-2,6-бисфосфат, наиболее мощный активатор гликолитического фермента фосфофруктокиназы-1 (PFK-1). Потеря PTEN снижает активность специфических для этих ферментов лигаз, чем стабилизирует их. Активация пути PI3К/Akt при мутантном PTEN обеспечивает фосфорилирование 6-фосфофрукто-2-киназы/фруктозо-1,6-бифосфатазы (PFKFB2) и активацию гликолитического фермента фосфофруктокиназы-2 (PFK-2). Также активный Аkt способствует экспрессии глюкозного транспортера (GluT1) на цитоплазматической мембране. Так утрата PTEN усиливает поглощение глюкозы и активность ферментов гликолиза, что крайне актуально для опухолевых клеток [6, 7].
Глутамин также используется злокачественными клетками для своих энергетических нужд. Потеря PTEN стабилизирует глутаминазу, которая запускает цикл глутаминолиза.
Аминокислоты с разветвленной боковой цепью (BCAA) — лейцин, изолейцин и валин — также могут быть использованы для синтеза белка или окислены в качестве источника энергии опухолевой ткани. Обнаружено, что катаболизм BCAA активируется в клетках рака предстательной железы с нокдауном PTEN. Предполагается, что ключевую роль в этом процессе играет мобилизация аминотрансферазы-1 с разветвленной цепью (BCAT1) — фермента, участвующего в первом этапе катаболизма BCAA [3].
Отсутствие экспрессии PTEN в клетках опухоли приводит к усилению синтеза жирных кислот de novo и их β-окислению, увеличению гликолиза и глутаминолиза, а также катаболизма аминокислот с разветвленной боковой цепью.
Напротив, у трансгенных мышей с модулированной сверхэкспрессией PTEN отмечено снижение поглощения клетками глюкозы и глутамина с одновременным усилением митохондриального окислительного фосфорилирования, что формирует устойчивость к онкогенной трансформации таких клеточных линий [8].
Метаболическое перепрограммирование, вызванное потерей экспрессии онкосупрессора PTEN, обеспечивает поставку биоматериалов и энергии для пролиферации злокачественных клеток и прогрессирующего роста опухоли. Восстановление гомеостатического баланса при мутациях PTEN представляет привлекательную терапевтическую мишень в таких опухолях.
Список литературы:
- Alwhaibi A.et al. Genome atlas analysis based profiling of Akt pathway genes in the early and advanced human prostate cancer. Oncoscience. 2019; 6(5-6):317-336.
- Shearn C.T. et al. Short term feeding of a high fat diet exerts an additive effect on hepatocellular damage and steatosis in liver-specific PTEN knockout mice. PLoS ONE.20144;9:e96553.
- Zhou X.et al. Effect of PTEN loss on metabolic reprogramming in prostate cancer cells. Oncol Lett. 2019;17(3):2856–2866.
- Chen C.Y., Chen J., He L., Stiles B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front Endocrinol (Lausanne). 2018;9:338.
- Debebe A. et al. Wnt/beta-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene.2017;36:6020–9.
- Shinde S.R., Maddika S. PTEN Regulates Glucose Transporter Recycling by Impairing SNX27 Retromer Assembly. Cell Rep. 2017 Nov 7;21(6):1655-1666.
- Haddadi N. et al. PTEN/PTENP1:Regulating the regulator of RTK-dependent PI3K/Akt signalling, new targets for cancer therapy. Mol Cancer. 2018;17(1):37.
- Garcia-Cao I.et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62.
