
Синдром Лея

Синдром Лея (СЛ) — это гетерогенное генетически обусловленное заболевание, относящееся к группе митохондриальных энцефаломиопатий с аутосомно-рецессивным или митохондриальным типом наследования и связанное с мутациями в генах, кодирующих полипептиды комплексов дыхательной цепи митохондрий, а также белков, принимающих участие в их сборке на внутренней поверхности митохондриальной мембраны [4].
Первые описания данного синдрома были даны Денисом Леем в 1951 году [1].
На заре открытия данного заболевания появились предположения, что его причиной является нарушение метаболизма, однако они не получили своего клинического подтверждения.
Доктор F. Hommes в 1968 году описал семьи, у представителей которых наблюдалось снижение активности пируваткарбоксилазы [2]. С 1975 года появились данные, что причиной СЛ может являться недостаточность пируватдегидрогеназного комплекса, а позже были обнаружены молекулярно-генетические нарушения. С развитием генетики и биохимии было выяснено, что изменения генов в мтДНК, которые ответственны за кодирование субъединицы АТФ-азы или тРНК — ядерных генов, кодирующих полипептиды комплекса дыхательной цепи митохондрий, — а также за нарушения в генах, отвечающих за сборку КДЦМ на митохондриальной мембране, приводят к развитию синдрома Лея [3]. Всего насчитывается 12 генов: NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, SDHA, SURF1, COX10, COX15, SCO2, BCS1L [1].
Чаще всего дефект обнаруживается в результате недостаточности IV КДЦМ-цитохром С-оксидазе (COX). Этот фермент является последним в электронно-транспортной системе митохондрий. СОХ включает в себя 13 субъединиц: 11 кодируются ядерными генами, 3 – мтДНК.
Чаще всего данная патология возникает в результате мутации гена SURF1, который лежит в хромосоме 9q34, кластере 6. Данный белок включен во внутреннюю мембрану митохондрии, и нарушения в нем приводят к синтезу укороченного белка и, как следствие, к повреждению СОХ-комплекса [4].
В результате биохимических исследований у большинства пациентов с СЛ обнаруживается повышение лактата в крови и спинномозговой жидкости. Повышение соотношения лактат/пируват является отражением нарушения окислительно-восстановительного баланса в цитоплазме [4].
Те пациенты, у которых мутации не превышают 70% от всех мтДНК к конкретной ткани, не имеют клинических проявлений данного синдрома. Однако при превышении данного порога могут проявляться симптомы, которые будут описаны ниже [1].
Синдром Лея чаще всего проявляется в детстве в виде утраты уже имеющихся психомоторных навыков, мозжечковыми и экстрапирамидными расстройствами, судорогами, мышечной гипотонией. Однако могут проявляться и такие симптомы как нистагм, потеря слуха, атрофия зрительного нерва. Дети отстают в своем развитии, отмечается регресс психомоторных функций, вялость, сонливость. Такие пациенты склонны к лактоацидозам. Ночью и при физических нагрузках возникают проблемы с дыханием в виде апноэ, диспноэ, тахипноэ [1].
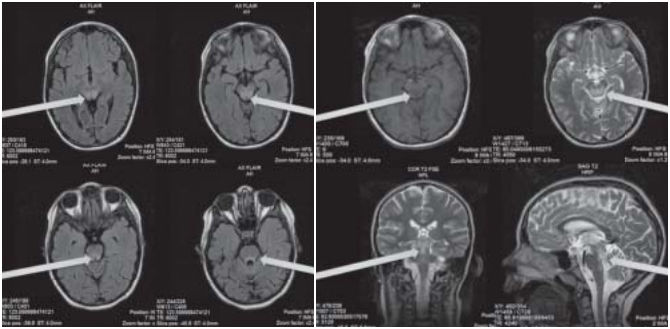
При МРТ-диагностике наблюдается снижение МР-сигнала в различных областях ГМ.
Также пациентам с синдромом Лея свойственна «парадоксальная гиперкетонемия» — повышение уровня кетоновых тел после пищевой нагрузки и высокое соотношение 3-гидроксибутират/ацетоацетат в крови. При проведении анализа органических кислот мочи может наблюдаться повышенная экскреция органических кислот, участвующих в цикле Кребса (фумаровая, янтарная и др.) [4].
Синдром Лея имеет следующую классификацию:
- типичный СЛ без эпилепсии и кардиомиопатии, обусловленный дефектом гена SURF1 (локус 9q34);
- Лей-подобный синдром, обусловленный мутацией гена SCO2, локус 22q13;
- СЛ с дистонией, тугоухостью и метилмалоновой ацидурией (дефект гена SUCLA2, локус 13q12.2-q13);
- СЛ с нефрозом или почечной недостаточностью (дефект биосинтеза субъединицы CoQ1, ген PDSS2, локус 6q21);
- Лей-подобный синдром с флюктуирующей периферической нейропатией и дистонией с лактатацидозом (дефект гена PDHA1, локус Xp22.2-p22.1);
- ювенильная форма СЛ с атрофией зрительных нервов (мутация гена TACO1, локус 17q22-q24.2) [1].
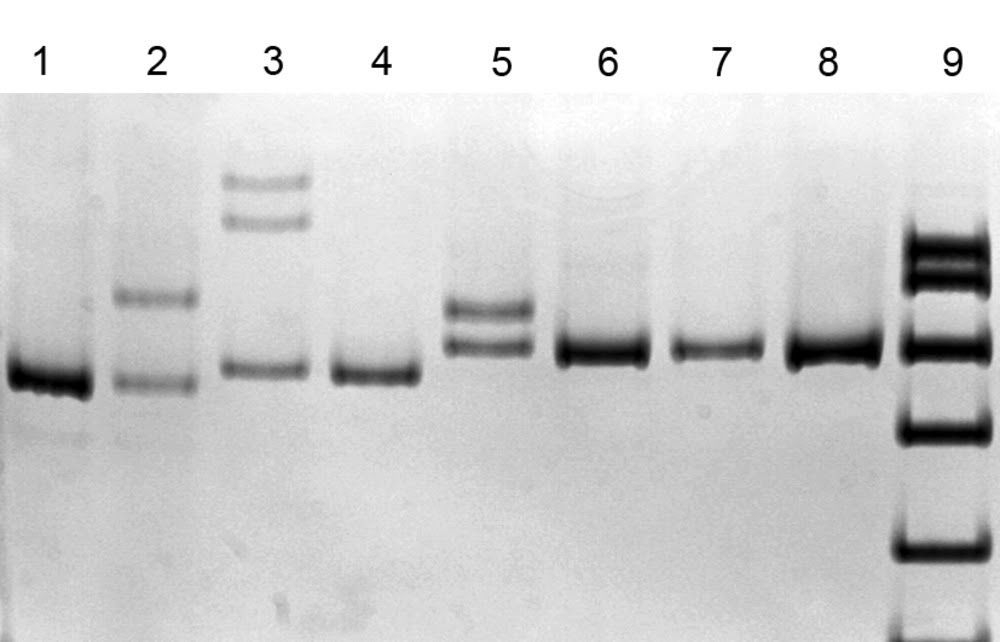
ДНК-диагностика 3 частых мутаций в гене SURF1 с помощью метода SSCP анализа (электрофорез в 8% полиакриламидном геле) [4].
.
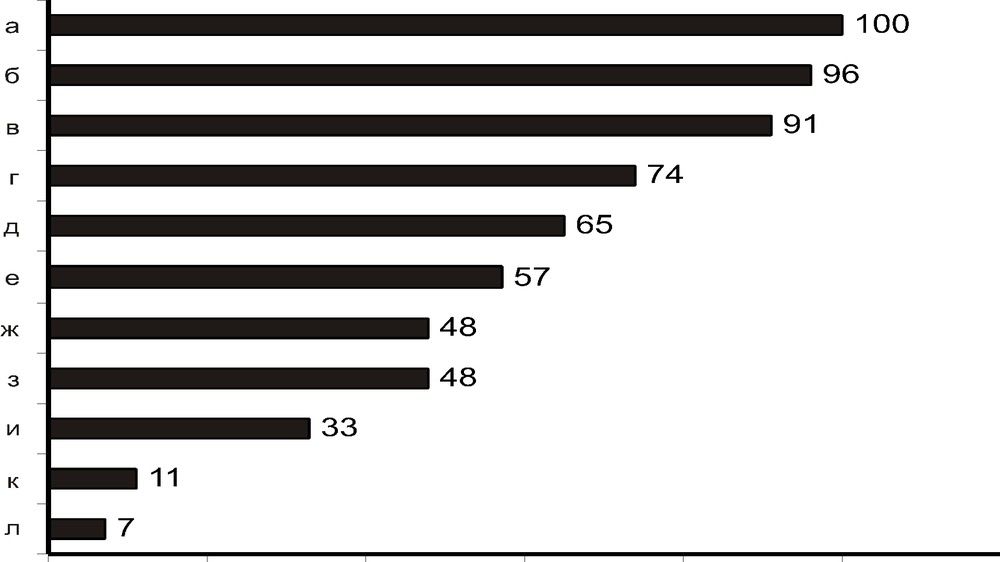
Статистика основных синдромов [4]:
- а — задержка психомоторного развития;
- б — мышечная гипотония;
- в — гипертрихоз;
- г — мозжечковый синдром;
- д — офтальмопарез;
- е — полинейропатический синдром;
- ж — дыхательные нарушения;
- з — пирамидный синдром;
- и — частичная атрофия зрительных нервов;
- к — экстрапирамидные расстройства;
- л — эпилептический синдром.
Источники:
- И. Г. Ковалев. Синдром Ли (подострая некротизирующая энцефаломиелопатия). Описание клинического случая. / И. Г. Ковалев, А. А. Соломасова, В. А. Чадаев, Э. Ю. Волкова, А. А. Холин, Н. Н. Заваденко // Вестник РГМУ. Неврология. — 2012 №2 — С. 31–35.
- Van Der Knaap M. S. / Valk J. Magnetic Resonance of Myelination and Myelin Disorders. // Berlin Springer Verlag — 2005 — С. 1084–1085.
- Hommes F., Leigh's encephalomyelopathy: an inborn error of gluconeogenesis. / Polman H., Reerink J // Arch Dis Child — 2008 — C. 423–426.
- П. Г. Цыганкова. Синдром Ли, обусловленный мутациями в гене SURF1: клинические и молекулярно-генетические особенности. / П. Г. Цыганкова, С. В. Михайлова, Н. А. Пичкур // Журнал неврологии и психиатрии им. С. С. Корсакова — 2010 — С. 25–32.
