
Нарушение выработки IFN-I и ответ воспалительных моноцитов и макрофагов вызывают летальную пневмонию у мышей, зараженных SARS-CoV

Ключевые моменты
— Коронавирус SARS-CoV вызывает летальную респираторную инфекцию у линии мышей BALB/c.
— Стойкая репликация SARS-CoV и отсроченная передача сигнала от IFN-I способствуют развитию заболевания.
— IFN-I вызывает миграцию патогенных воспалительных моноцитов и повышение проницаемости кровеносных сосудов.
— Степень тяжести заболевания снижается при отсутствии передачи сигнала от IFN-I.
Теоретическое обоснование
Высокопатогенные возбудители респираторных коронавирусных инфекций человека вызывают острое летальное заболевание, которое характеризуется избыточной воспалительной реакцией и повреждением легких. Однако факторы, приводящие к патологии легких, остаются не совсем изученными. На мышах, зараженных коронавирусом тяжелого острого респираторного синдрома (SARS-CoV), авторы демонстрируют, что стойкая репликация вирусов сопровождается отсроченной передачей сигнала от интерферона I типа (IFN-I), который контролирует воспалительные реакции и иммунопатологические процессы в легких при снижении выживаемости.
IFN-I обнаруживается до тех пор, пока титры вирусов не достигнут пика, однако раннее введение IFN-I улучшает течение патологических иммунных реакций. Отсроченный сигналинг IFN-I способствует накоплению патогенных воспалительных моноцитов и макрофагов, что приводит к повышению уровня цитокинов/хемокинов, увеличению проницаемости кровеносных сосудов, нарушению механизма вирус-специфического Т-клеточного ответа. Модифицирование ДНК с целью нарушения экспрессии гена рецепторов IFN-αβ (IFNAR) или деплеция воспалительных моноцитов и макрофагов защищает мышей от летальной инфекции, без влияния на вирусную нагрузку. Эти результаты показывают, что IFN-I и воспалительные моноциты и макрофаги способствуют развитию летальной инфекции, вызванной SARS-CoV, и являются потенциальными терапевтическими мишенями при лечении пациентов, зараженных коронавирусом и, возможно, иными респираторными вирусами.
Результаты
Ингибирование передачи сигнала от IFN-I снижает заболеваемость и летальность у мышей линии BALB/c, инфицированных SARS-CoV
Чтобы изучить, усугубляет ли передача сигнала от IFN-I течение тяжелого острого респираторного синдрома (ТОРС), авторы инфицировали мышей линии BALB/c в возрасте 7–9 недель, а также мышей, нокаутных по гену рецептора IFN-αβ (далее эта линия обозначается Ifnar⁻ᐟ⁻) летальной дозой SARS-CoV (3×10⁴ БОЕ). Отслеживалась тяжесть заболевания. У мышей линии BALB/c наблюдались: сильная вялость, взъерошенная шерсть, сгорбленная поза и затрудненное дыхание уже на третий день после заражения; все это сопровождалось прогрессирующим снижением веса. Примерно 85 % мышей умерли на восьмой день (рис. 1А–1С). Однако мыши линии Ifnar⁻ᐟ⁻ пережили заражение, имея лишь умеренную (приблизительно 15 %) потерю веса и легкую и умеренную клиническую форму болезни (рис. 1A–1C). Даже при более высоких дозах вируса (10⁵ БОЕ) все мыши Ifnar⁻ᐟ⁻ выжили. Мыши линии BALB/c среднего возраста (8–9 месяцев) высоко восприимчивы к SARS-CoV, но даже у этих мышей выживаемость увеличивалась при отсутствии передачи сигнала от IFN-I.
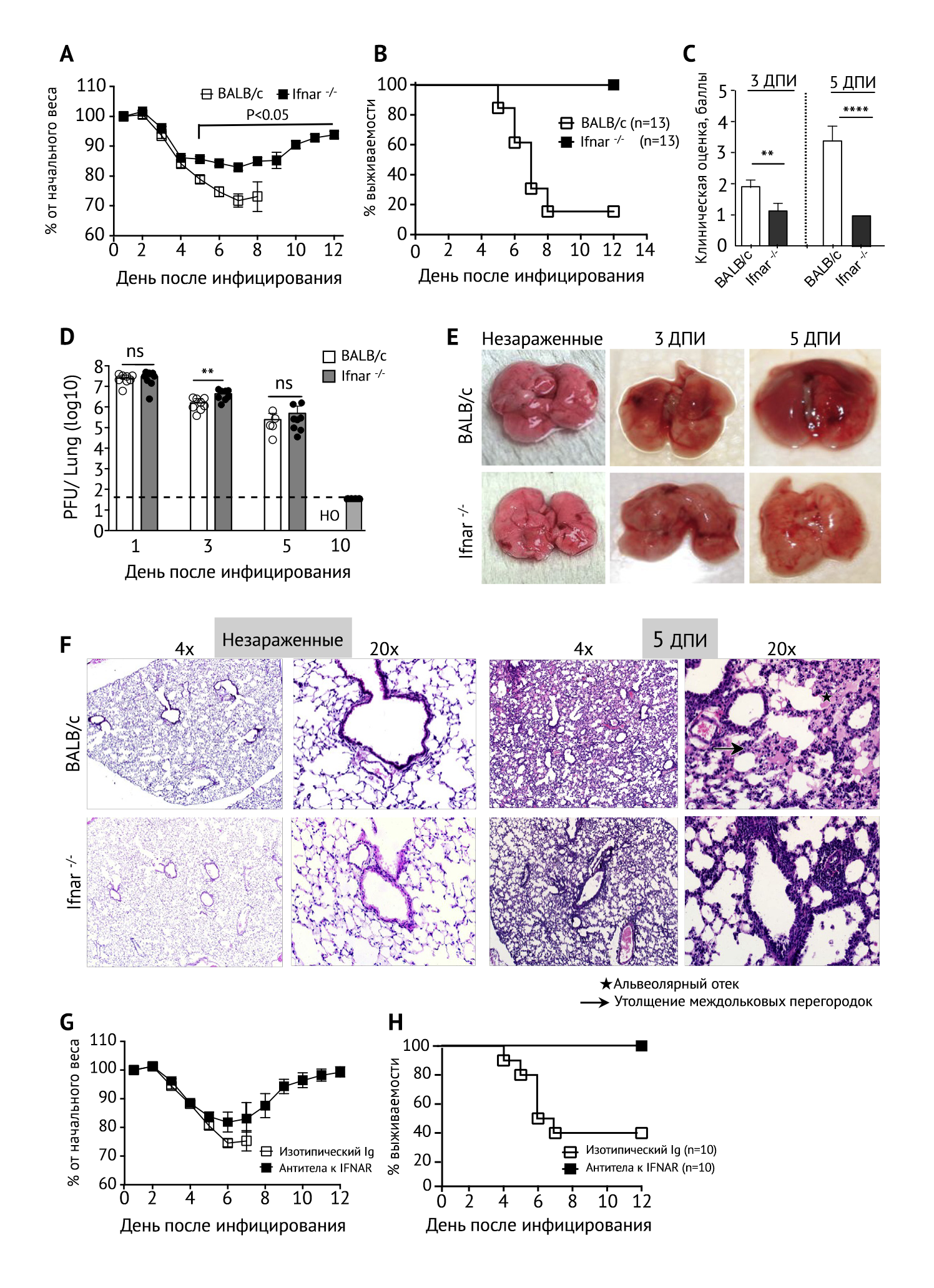
С. Клинические показатели, определяемые на 3 и 5 день после инфицирования.
D. Титры SARS-CoV в легких, определяемые с помощью анализа бляшкообразующих единиц (н/о — не определено).
Е. Макропрепараты легких на 3 и 5 дни после инфицирования.
F. Микроскопические изменения в легких незараженных мышей и на 5 день после инфицирования.
G и H. Кривые веса (G) и выживаемости (H) мышей, зараженных SARS-CoV, получавших антитела к рецептору IFNAR и контрольные изотипические антитела (MOPC21).
Данные являются репрезентативными для 2–3 независимых экспериментов с 4–5 мышами в группе.
Данные в (А), (С) и (D) представлены ± стандартная ошибка среднего (SEM).
*p < 0,05, **p < 0,01, и ***p < 0,001.
Результаты гистопатологических исследований и исследований макропрепаратов получены от 4–5 мышей в группе.
Суммарные вирусные нагрузки в тканях легких были одинаковыми у молодых мышей BALB/c и Ifnar⁻ᐟ⁻ во все дни после заражения, за исключением небольшого увеличения на третий день у Ifnar⁻ᐟ⁻ (рис. 1D). SARS-CoV был полностью выведен из легких этих мышей к десятому дню (рис. 1D). Общее обследование легких показало обширную гиперемию и венозное полнокровие в легких у группы мышей BALB/c на третий и пятый день, в то время как у группы Ifnar⁻ᐟ⁻ результаты были почти что нормальными (рис. 1E). Гистопатологическое исследование легких показало, что выраженный альвеолярный отек, десквамация бронхиального эпителия и утолщение междольковых перегородок в легких у мышей BALB/c наблюдались на пятый день, в то время как в группе Ifnar⁻ᐟ⁻ альвеолярный отек сопровождался минимальной перибронхиальной и периваскулярной лимфоплазмоцитарной инфильтрацией (рис. 1F). Острое повреждение легких, ассоциированное с ТОРС, часто сопровождается накоплением богатой белками жидкости в периваскулярном и альвеолярном пространствах (Franks с соавт., 2003; Gu с соавт., 2005; Nicholls с соавт., 2003). Для оценки повышения проницаемости сосудов, авторы оценили экстравазацию синего красителя Эванса в легких на пятый день. У мышей BALB/c, зараженных SARS-CoV, проницаемость сосудов в легких была значительно выше, однако у Ifnar⁻ᐟ⁻ она была гораздо ниже.
Поскольку эти результаты предполагают значимую роль передачи сигнала от IFN-I при тяжелом течении заболевания, аномалии развития также могут привести к улучшению результатов при отсутствии передачи сигнала от IFNAR. Чтобы исключить эту возможность, исследователи либо блокировали рецепторы IFN-αβ (IFNAR) у мышей линии BALB/c, либо давали им вместо лечения изотипические контрольные антитела до и после заражения SARS-CoV. В соответствии с проявлениями болезни у линии Ifnar⁻ᐟ⁻, лечение антителами к IFNAR значительно сократило потерю веса и повысило выживаемость по сравнению с мышами, которые получали контрольные антитела (рис. 1G и 1H).
Для определения общего характера вышеуказанных результатов авторы инфицировали линии BALB/c и Ifnar⁻ᐟ⁻ вирусом гепатита мышей (MHV-1), вирусом гриппа A (IAV, штамм PR8) и двумя РНК-вирусами, которые также вызывают у мышей острые респираторные заболевания. В соответствии с результатами предыдущих исследований (Cervantes-Barragan с соавт., 2009; Seo с соавт., 2011), передача сигнала от IFN имеет критическое значение для защиты от инфекции: все мыши линии Ifnar⁻ᐟ⁻ погибли от полулетальной инфекции MHV-1 и штамма гриппа PR8, в то время как только приблизительно 10 % MHV-1 и 20 % PR8-инфицированных мышей BALB/c, соответственно, умерли. Эти результаты свидетельствуют, что существуют уникальные особенности инфекции, вызываемой SARS-CoV, из-за которых передача сигнала от IFN-I является скорее патологической, а не защитной.
Быстрая репликация SARS-CoV и отсроченная экспрессия IFN-I в легких
Результаты предыдущих исследований (Frieman и соавт., 2010; Roberts и соавт., 2007; Zhao и соавт., 2009) и результаты, показанные на рис. 1D, демонстрируют быструю скорость репликации SARS-CoV в легких. Как показано на рис. 2A, титры вируса практически максимальны через 16 часов первого дня после заражения. Иммуногистохимическое исследование легких, инфицированных SARS-CoV, выявило репликацию вируса в дыхательных путях и паренхиме легких. Окрашивание вирусного антигена обнаруживалось в пневмоцитах II типа через 16 часов первого дня (рис. 2B). К 24 и 48 часам были обнаружены вирусные антигены, распределенные по дыхательным путям и паренхиме легких, с более интенсивным распространением антигенов в пневмоцитах II типа и с менее — в пневмоцитах I типа (рис. 2B), аналогично результатам исследования биоптатов людей, зараженных SARS-CoV (Gu и соавт., 2005; Nicholls и соавт., 2003). Распределение и количество клеток, содержащих вирусные антигены, были одинаковыми у мышей линий BALB/c и Ifnar⁻ᐟ⁻ в соответствии с титрами вируса, показанными на рис. 2A.
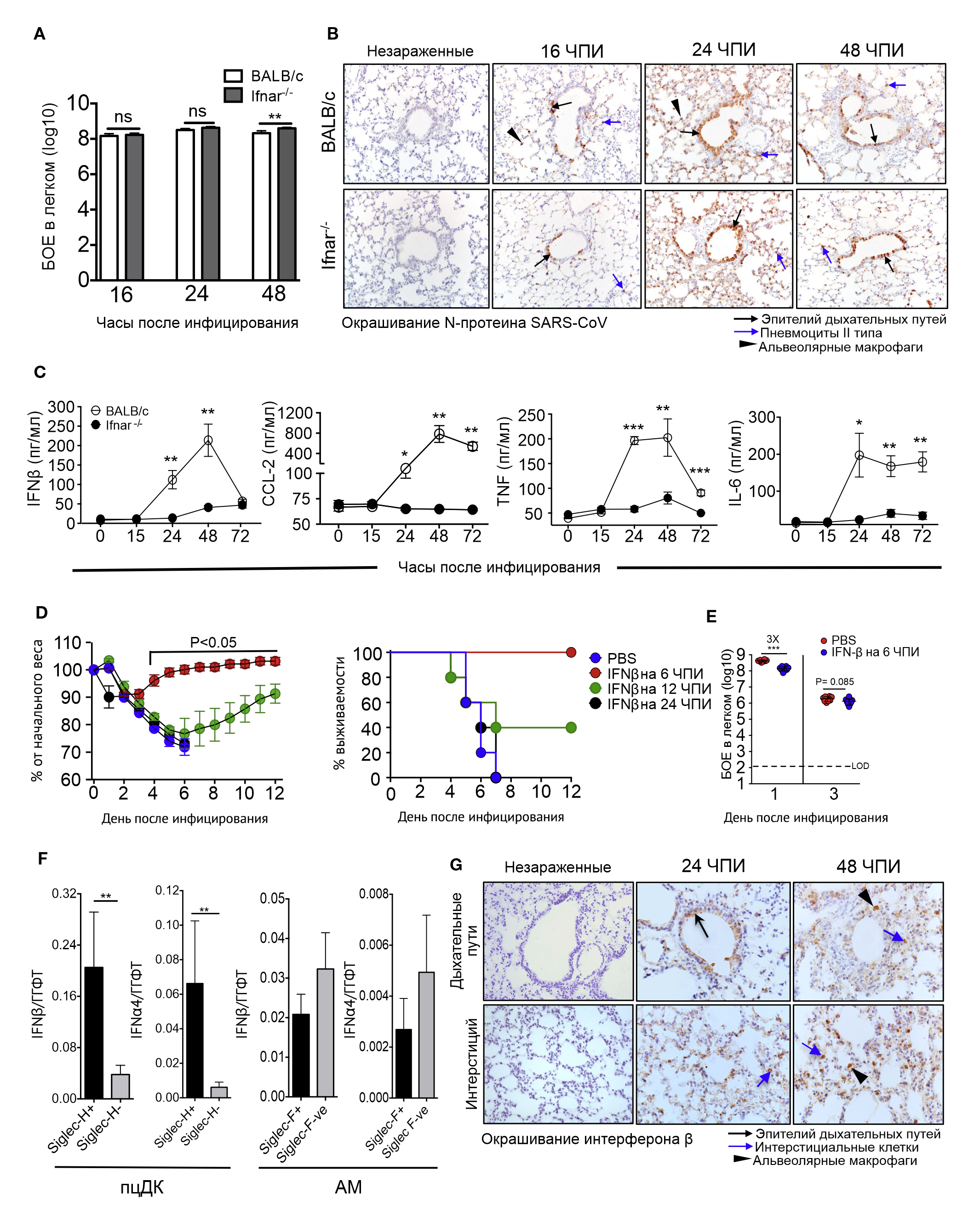
B Иммуногистохимическое исследование N-протеина SARS-CoV в разное время после заражения.
C Уровни цитокинов/хемокинов в БАЛЖ мышей линий BALB/c и Ifnar −/− в разное время после инфицирования.
D Кривые веса и выживаемости после терапии IFN-β (2000 U, интраназально, разовая доза в 6, 12 или 24 час после инфицирования).
E Вирусная нагрузка в легких в 1 и 3 день после инфицирования у мышей, получавших IFN-β (2000 U, 6 часов после инфицирования) или натрий-фосфатный буфер (phosphate buffered saline, PBS).
F Легочные клетки, изъятые у мышей на 24 час после инфицирования SARS-CoV, были отсортированы с помощью метода иммуномагнитной сепарации (MACS) на Siglec-H-положительные и отрицательные и на Siglec-F-положительные и отрицательные клетки.
Отобранные клетки анализировали на уровни мРНК IFN-α4 и IFN-β.
G Иммуногистохимическое исследование экспрессии IFN-β в дыхательных путях легких (верхняя панель) и паренхимы легких (нижняя панель) в разное время после инфицирования (С).
Данные (А) и (С)-(Е) получены в результате двух независимых экспериментов, 4–5 мышей в группе на 1 эксперимент. Данные в пунктах (А) и (С)-(F) представлены ± стандартная ошибка среднего (SEM).
**p < 0,01, ***p < 0,001.
IFN-I важен для запуска защитного иммунного ответа, но он не был обнаружен в бронхоальвеолярной лаважной жидкости мышей (БАЛЖ) группы BALB/c до 24 часов первого дня после заражения. Аналогичным образом, другие цитокины и хемокины, вовлеченные в иммунный ответ, также показали отсроченную экспрессию при анализе БАЛЖ (рис. 2C). Напротив, у линии Ifnar⁻ᐟ⁻ значительно снизился уровень цитокинов и хемокинов в БАЛЖ по сравнению с линией BALB/c (рис. 2C). Поскольку отсроченная экспрессия медиаторов воспаления может способствовать избыточной реакции клеток врожденного иммунитета, далее авторы изучили, может ли введение IFN-I до пика вирусной репликации изменить клинические проявления и увеличить выживаемость. С этой целью исследователи вводили рекомбинантный IFN-β (2000 U) интраназально в разное время после заражения. В соответствии с предыдущими исследованиями (Kumaki с соавт., 2011) IFN-I, введенный на 6 час первого дня до пика репликации вируса, но не на 24 час первого дня, полностью защищает мышей от потери веса и развития заболевания. Введение IFN-β на 12 час первого дня приводит к промежуточному уровню защиты. (рис. 2D), а при введении на 6 час умеренно снижает вирусные титры в легких в первый день (рис. 2E).
Далее авторы исследовали клеточный источник IFN-I в легких, инфицированных SARS-CoV. Поскольку плазмоцитоидные дендритные клетки (пцДК) и альвеолярные макрофаги (АМ), как известно, являются важными источниками IFN-I при заболеваниях, ассоциированных с РНК-содержащими вирусами (Killip с соавт., 2015), исследователи предварительно очистили ткани от пцДК и альвеолярных макрофагов, имеющих поверхностные белки Siglec-H и Siglec-F соответственно (метод магнитной сепарации). Затем уровни мРНК IFN-β и IFN-α4 измерены методом ПЦР в реальном времени с обратной транскрипцией. Белок Siglec-H экспрессируется пцДК и субпопуляциями макрофагов в лимфоидных тканях, но главным образом, легочными пцДК (Swiecki и соавт., 2014). Очищенные плазмоцитоидные ДК инфицированных мышей демонстрировали значительно более высокие уровни мРНК IFN-β и IFN-α4 в сравнении с клетками, которые не содержат белок Siglec-H (рис. 2F). В соответствии с этими результатами, человеческие пцДК также являются постоянными источниками IFN после заражения SARS-CoV in vitro (Cervantes-Barragan и соавт., 2007). Альвеолярные макрофаги также вырабатывали IFN-I, но не более, чем другие клетки. Кроме того, авторы также окрашивали участки тканей на IFN-β для оценки содержания IFN-I в других легочных клетках и обнаружили экспрессию в клетках дыхательных путей и интерстициальных клетках (рис. 2G). В совокупности эти результаты позволяют предположить, что многие типы клеток, но особенно пцДК, экспрессируют IFN-I при заражении вирусом SARS-CoV у мышей и, скорее всего, у людей.
Сигналинг IFN-I регулирует миграцию и активацию моноцитов и макрофагов. Затем исследователи оценили, способствовал ли IFN-I миграции клеток врожденного иммунитета у мышей, зараженных SARS-CoV. Общее количество нейтрофилов, альвеолярных макрофагов, естественных киллеров и пцДК в легких сравнили у мышей, зараженных SARS-CoV, у линий BALB/c и Ifnar⁻ᐟ⁻ в первый и третий день. Произошло значительное увеличение количества клеток, содержащих лимфоцитарный антиген Ly6С в большом количестве и интегрин CD11b (далее этот клеточный фенотип обозначаются Ly6ChiCD11b+), в легких мышей популяции BALB/c, и это увеличение прекращалось при отсутствии передачи сигнала от IFN-I (рис. 3A и 3B). На третий день произошло примерно шестикратное увеличение инфильтрации клетками Ly6ChiCD11b+ легочной ткани у мышей BALB/c по сравнению с мышами Ifnar⁻ᐟ⁻ (рис. 3B). Фенотипическое исследование показало, что эти клетки экспрессировали CCR2 (рецептор β-хемокинов), F4/80 (макрофагальный маркерный протеин) и CD11, но не белок Ly6G и лектин Siglec-Н. Это значит, что эти клетки были моноцитами и макрофагами (рис. 3С). Кроме того, уровни CD69 (раннего антигена активации T-лимфоцитов), BST-2 (тетерина), белков CD80 и CD86 были значительно выше на поверхности моноцитов и макрофагов, взятых из легких мышей BALB/c в сравнении с мышами линии Ifnar⁻ᐟ⁻ (рис. 3D), что соответствует большей активации клеток. Хемокины CCL2, CCL7 и CCL12, которые являются лигандами для рецептора CCR2, также как и ГМ-КСФ (гранулоцитарно-макрофагальный колониестимулирующий фактор), участвуют в миграции воспалительных макрофагов и моноцитов (Shi and Pamer, 2011; Song и соавт., 2015). В то время как экспрессия CCR2 в легких мышей Ifnar⁻ᐟ⁻ значительно уменьшалась в течение 24 и 72 часов после заражения в сравнении с мышами BALB/c, экспрессия ГМ-КСФ замедлилась на 16 час. При прямом измерении ex vivo CCL2 продуцировался преимущественно моноцитами и макрофагами, а передача сигнала от IFN-I была достаточной для того, чтобы вызвать экспрессию лиганда к CCR2 клетками костного мозга. Таким образом, сигналинг IFN-I способствовал накоплению высокоактивированных воспалительных моноцитов и макрофагов в легких, зараженных SARS-CoV, и этот процесс усиливался моноцитами и макрофагами посредством выработки лигандов к CCR2.
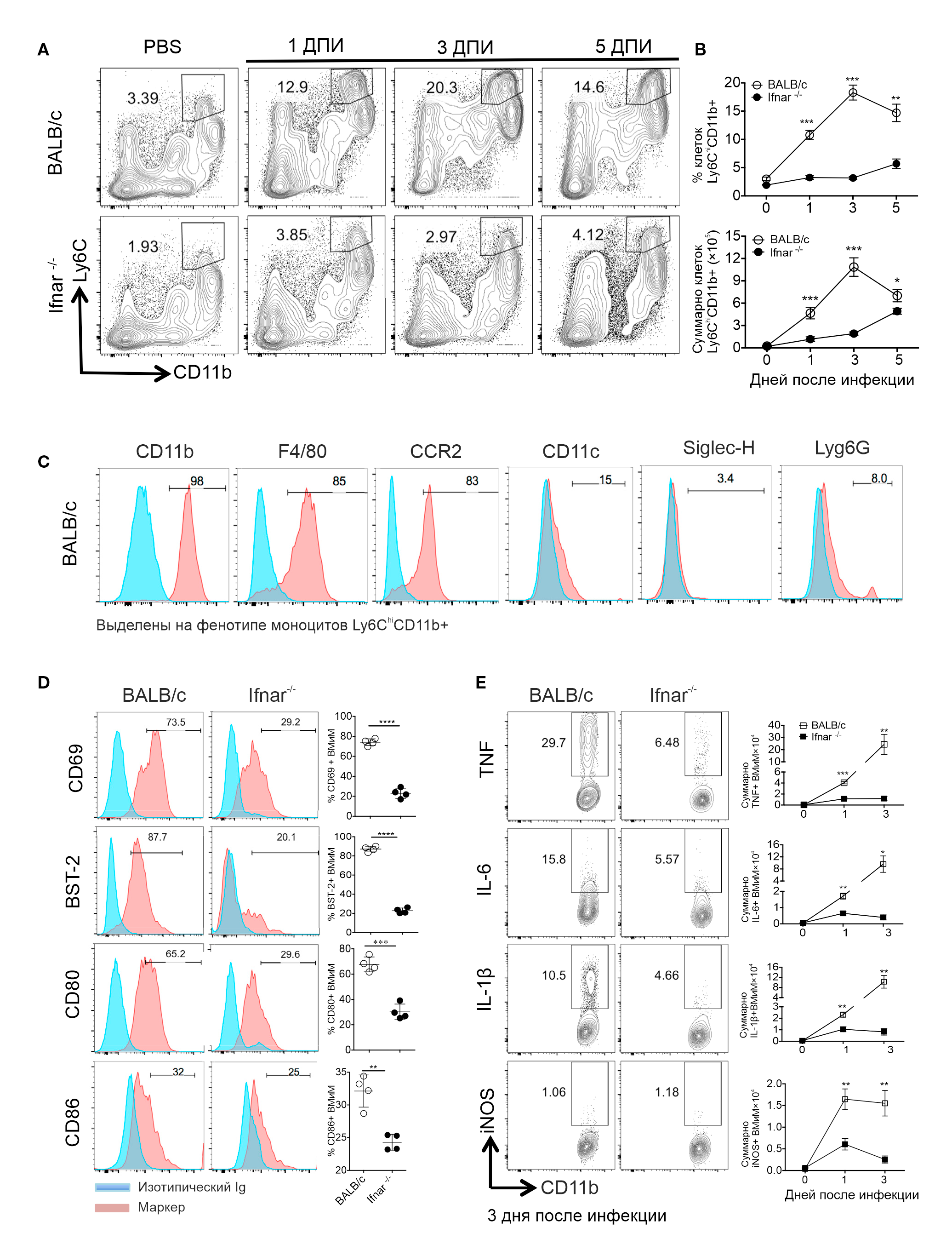
В Процент и общее число клеток Ly6ChiCD11b+ в легких.
С Экспрессия фенотипического маркера на воспалительных моноцитах из легких мышей BALB/c на 3 день после инфицирования.
D Уровни маркеров активации клеток на поверхности макрофагов и моноцитов легких на 3 день.
E Общее количество цитокин-положительных макрофагов и моноцитов в легких, определенное после 7-часовой инкубации ex vivo c добавлением брефельдина A.
Данные получены из 2–3 независимых экспериментов с 4 мышами в группе на 1 эксперимент.
Данные в (В), (D) и (Е) представлены ± стандартная ошибка среднего (SEM).
*p < 0,05, **p < 0,01, ***p <0,001.
ДПИ — день после инфицирования
TNF — фактор некроза опухоли
ВМиМ — воспалительные моноциты и макрофаги
Чтобы определить, были ли моноциты и макрофаги также ответственны за повышенную экспрессию провоспалительных цитокинов, были окрашены моноциты и макрофаги мышей BALB/c и Ifnar⁻ᐟ⁻ ex vivo на наличие внутриклеточных TNF, IL-6, IL-1-β и NO-синтаз. В результате были выявлены значительно более высокие процентные доли и числа клеток, выделявших эти провоспалительные цитокины в легких мышей BALB/c по сравнению с Ifnar⁻ᐟ⁻ (рис. 3E). Далее авторы оценили, являются ли моноциты и макрофаги главным источником этих цитокинов в зараженных легких путем деплеции клеток с помощью моноклонального антитела к рецептору CCR2 (мАТ МС21), поскольку CCR2 экспрессируется в особенности при высоких концентрациях провоспалительных моноцитов (Mack с соавт., 2001). Антитело MC21 обеспечивает специфическую деплецию макрофагов и моноцитов фенотипа Ly6ChiCD11b+, снижая их уровень у мышей линии Ifnar⁻ᐟ⁻. Лечение моноклональными антителами МС21 значительно снижает уровни CCL2, TNF, и IL-6 в бронхоальвеолярной лаважной жидкости. Это напрямую подтверждает, что моноциты и макрофаги являются главным источником цитокинов/хемокинов в инфицированных легких.
Деплеция моноцитов и макрофагов улучшает течение заболевания, вызванного вирусом SARS-CoV
Для непосредственного установления связи между повышенной инфильтрацией легочной ткани макрофагами и моноцитами и иммунопатологией легких, вызванной SARS-CoV, исследователи вызвали деплецию этих клеток с помощью моноклонального антитела MС21. Этот метод лечения защитил от летального исхода при инфекции, что подтвердило решающую роль моноцитов и макрофагов в повышении заболеваемости и смертности, индуцированных вирусом SARS-CoV (рис. 4А). Для подтверждения этих результатов еще одной когорте мышей было введено антитело против белка BST-2, так как он на высоком уровне экспрессировался активированными моноцитами и макрофагами (рис. 3D). BST-2 конститутивно экспрессируется на плазмоцитоидных дендритных клетках и его количество увеличивается на моноцитах, макрофагах и некоторых стромальных клетках при наличии воспалительных стимулов (Blasius и соавт., 2006). Деплеция BST-2 полностью защищает мышей линии BALB/c от летального исхода, в то время как около 70 % мышей группы, получавшей лечение контрольными иммуноглобулинами, умерли (рис. 4С). Деплеция моноцитов и макрофагов или введение антител минимально повлияли на титры вируса в легких (приблизительно в 2 раза) (рис. 4B и 4D). Гистологическое исследование легких, обработанных MC21 или моноклональными антителами к BST-2, показало уменьшение альвеолярного отека и десквамации бронхиального эпителия без изменения клеточной инфильтрации, по сравнению с группами мышей, которые получали контрольный иммуноглобулин (рис. 4E). Кроме того, деплеция моноцитов и макрофагов также уменьшает проницаемость сосудистой стенки. Примечательно, что терапия антителами MC21 также уменьшила количество пцДК в легких, даже если они не экспрессировали рецептор CCR2, возможно, за счет уменьшения выработки хемокинов. Как отмечалось выше, моноциты и макрофаги экспрессируют высокие уровни воспалительных цитокинов (рис. 3E), что приводит к повышению их уровня в легких (рис. 2C), что также может способствовать более тяжелому течения заболевания у мышей линии BALB/c. В соответствии с этим, нейтрализация одного провоспалительного медиатора, TNF, который был наиболее повышен у группы мышей BALB/c в сравнении с группой Ifnar⁻ᐟ⁻ (рис. 3E), обеспечила частичную, но, тем не менее, существенную защиту по сравнению с контрольной группой мышей (рис. 4F). Хотя эти результаты демонстрируют ключевую роль моноцитов и макрофагов при тяжелой форме заболевания у мышей, инфицированных SARS-CoV, нейтрофилы часто наносят вред при респираторных вирусных инфекциях (Brandes и соавт., 2013). Однако, как отмечалось выше, у мышей линии BALB/c немного выше количество нейтрофилов немного выше и их деплеция не повлияла на выживаемость (рис. 4G). В совокупности эти результаты демонстрируют роль моноцитов и макрофагов на нескольких стадиях воспалительного процесса, что приводит к иммунопатологическим изменениям у мышей, зараженных SARS-CoV.
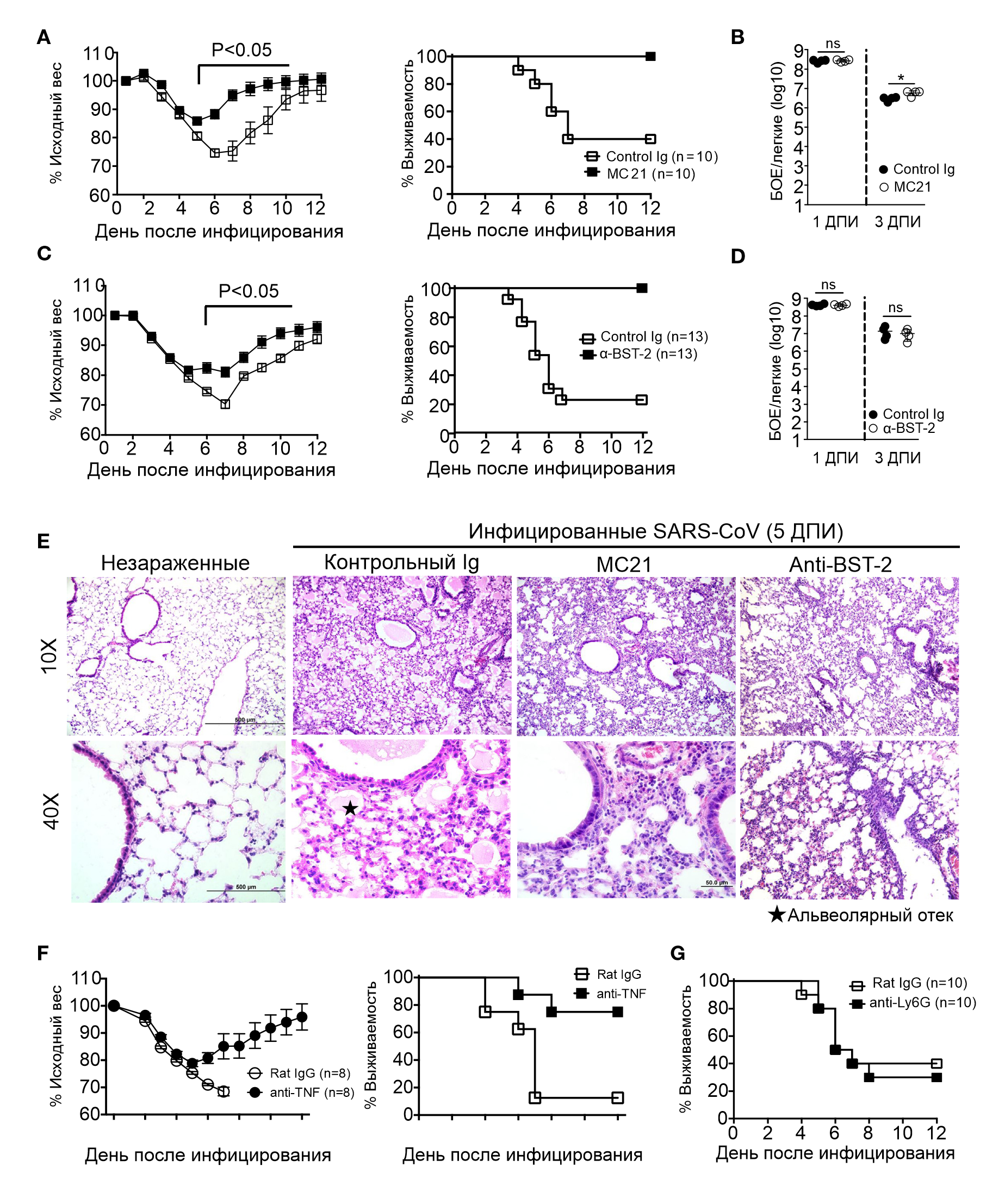
A и C. Кривые веса и выживаемости мышей, инфицированных SARS-CoV, после терапии контрольными антителами, MC21 (A) или антителами к BST-2 (C).
B и D. Вирусные титры в легких в 1 и 3 день после контрольного Ig и MC21 (B) или лечение моноклональными антителами к BST-2 (D).
E. Гистологические изменения в легких незараженных, получавших лечение изотипическими антителами, деплецией макрофагов и моноцитов у мышей линии BALB/c, зараженных SARS-CoV, на 5-й день.
F. Кривые веса и выживаемости контрольной группы и группы BALB/c, получавших антитела к TNF-α
G. Выживаемость мышей BALB/c, инфицированных SARS-CoV, после деплеции нейтрофилов.
Данные являются репрезентативными для 2 независимых экспериментов (4–5 мышей в группе на 1 эксперимент).
Данные в (A)–(D) представлены ± стандартная ошибка среднего (SEM).
*p < 0,05, **p < 0,01,***p < 0,001.
Передача сигнала от IFN-I к гемопоэтическим клеткам усиливает течение болезни у мышей BALB/c, инфицированных SARS-CoV
Выявив ключевую роль ВМиМ в усугублении тяжести SARS-CoV, авторы оценили нужен ли сигналинг IFN-I к гемопоэтическим и не гемопоэтическим клеткам для инициации миграции ВМиМ к месту инфекции.
Для решения этой задачи авторы создали химеры костного мозга между разрозненными CD45 у мышей BALB/c и Ifnar⁻ᐟ⁻. Приживление донорских клеток было подтверждено проточным цитометрическим анализом экспрессии CD45.1 и CD45.2 на лейкоцитах периферической крови через 5 недель после пересадки (рис. 5А). Через 6 недель после восстановления костного мозга химерные мыши столкнулись с летальной дозой SARS-CoV. Только 10 % мышей в группе Ifnar⁻ᐟ⁻→ Ifnar⁻ᐟ⁻ и 20 % мышей в группе Ifnar⁻ᐟ⁻→BALB/c скончались. Напротив, 100 % мышей группы BALB/c→BALB/c и ∼70 % мышей-химер группы BALB/c→ Ifnar⁻ᐟ⁻ погибли от SARS-CoV (рис. 5B). Относительная восприимчивость мышей с химерным костным мозгом к инфекции SARS-CoV коррелировала с количеством ВМиМ, проникающих в легкие на 3 день заражения (рис. 5C и 5D). Таким образом, у мышей BALB/c с восстановленным костным мозгом, наблюдалась повышенная инфильтрация ВМиМ, тогда как мыши, получившие Ifnar⁻ᐟ⁻ костный мозг, в основном были устойчивы к SARS-CoV (рис. 5С и 5D). Вместе эти результаты показывают, что сигналинг от IFN-I, в основном к кроветворным клеткам, способствовал развитию иммунопатологии легких у мышей BALB/c, зараженных SARS-CoV.
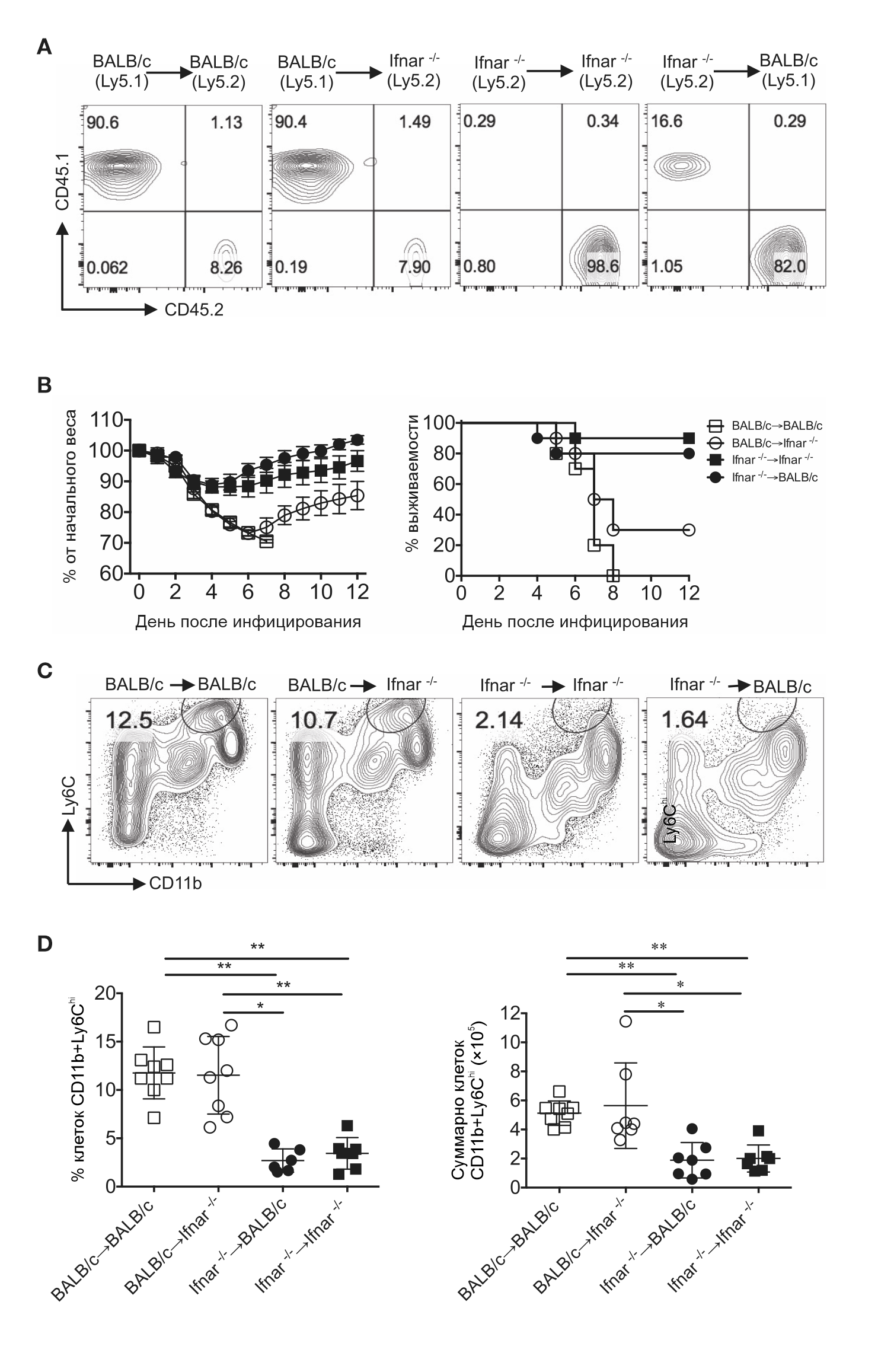
В. Выживаемость химерных мышей отслеживалась после заражения SARS-CoV (103 БОЕ интраназально, 4–5 мышей в группе, 2 независимых эксперимента).
С. Анализ инфильтрации суспензии легочных клеток химерных мышей, инфицированных SARS-CoV, воспалительными моноцитами и макрофагами на третий день после заражения.
D. Проценты и общее число воспалительных моноцитов и макрофагов на третий день после инфицирования.
Для (С) и (D) данные репрезентативны для 2–3 независимых экспериментов (2–3 мыши в группе на эксперимент). Данные в (D) представлены в виде ± стандартная ошибка среднего (SEM).
*p < 0,05, **p < 0,01, и ***p < 0,001.
IFN-I-опосредованные воспалительные реакции нарушают вирус-специфический T-клеточный ответ у мышей BALB/c, зараженных SARS-CoV
Так как для элиминации SARS-CoV необходим устойчивый Т-клеточный ответ (Channappanavar и соавт., 2014; Zhao и соавт., 2010), была проведена его оценка на шестой день у зараженных мышей групп BALB/c, Ifnar⁻ᐟ⁻ и у тех мышей, которым вводили моноклональное антитело MC21. Общее количество вирус-специфических Т-клеток двух субпопуляций — CD8 (эпитоп S366) и CD4 (эпитоп N353) — в легких было значительно меньше у мышей группы BALB/c по сравнению с группой Ifnar⁻ᐟ⁻ или теми мышами, что проходили лечение антителом MC21 (рис. 6A-6D). Предыдущие исследования ассоциировали субоптимальный Т-клеточный ответ с нарушением миграции дендритных клеток дыхательных путей (ДКДП) в лимфатические узлы для дальнейшей антиген-специфической пролиферации (Zhao и соавт., 2011, 2009). Поэтому для изучения того, способствовало ли уменьшение миграции дендритных клеток субоптимальному вирус-специфическому Т-клеточному ответу, подопытным животным обеих групп (BALB/c и Ifnar⁻ᐟ⁻) за 6 часов до инфицирования SARS-CoV интраназально вводили 6-карбоксифлуоресцеин сукцинимидиловый эфир (6-Carboxyfluorescein succinimidyl ester, CFSE). Проанализирована миграция дендритных клеток в лимфатические узлы на 18 час после заражения. Несмотря на то, что процент мигрировавших CFSE-положительных клеток был ниже у мышей Ifnar⁻ᐟ⁻, было получено сходное число таких клеток и в группе мышей BALB/c, и в группе Ifnar⁻ᐟ⁻ (рис. 6E и 6F). Эти результаты свидетельствуют, что нарушения Т-клеточного ответа не влияют на слабую миграцию дендритных клеток дыхательных путей.
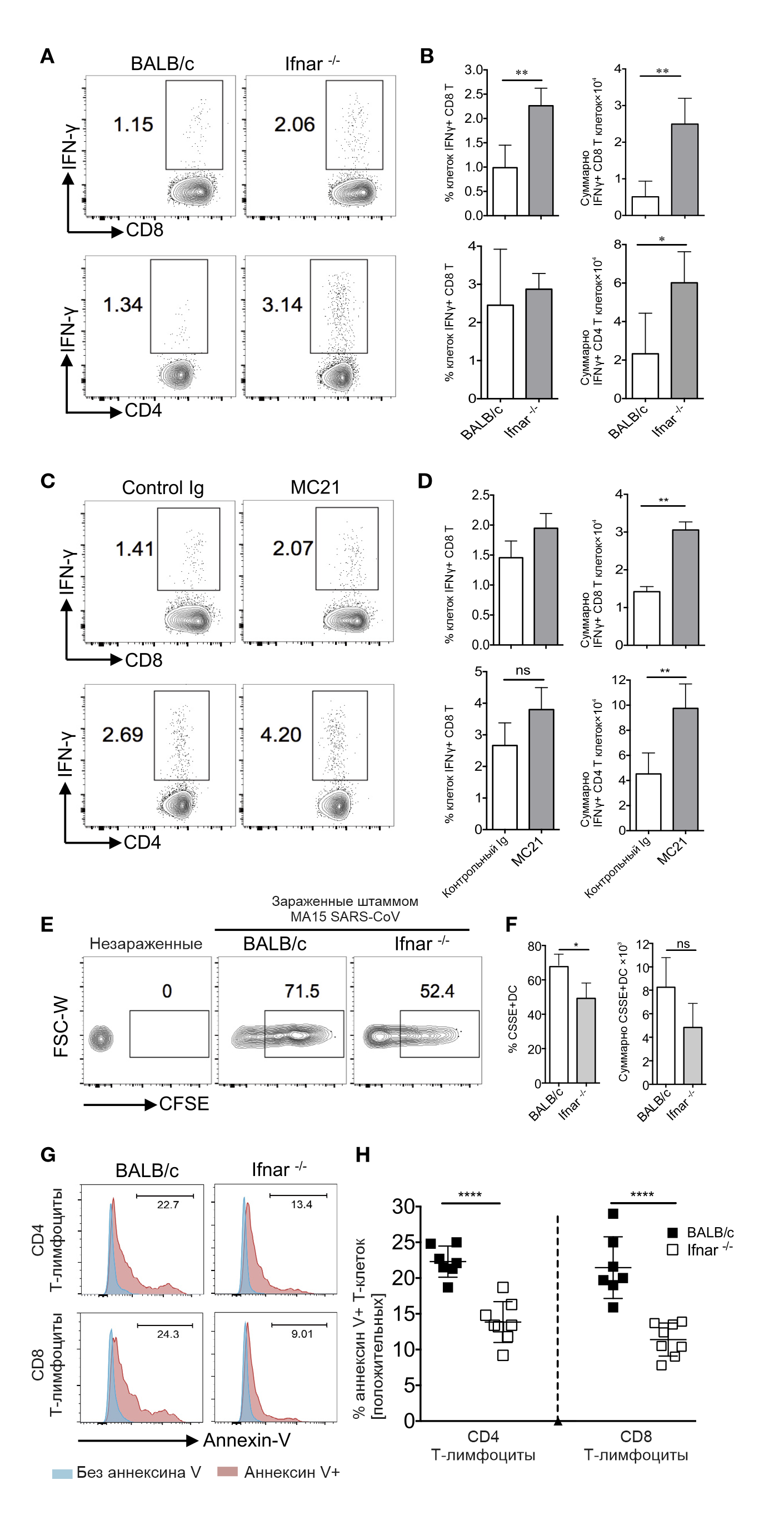
A и B. Графики FACS (A) и гистограммы (B) показывают процентное и общее число вирус-специфических CD4 и CD8 Т-клеток в легких мышей группы BALB/c и Ifnar −/−.
С и D. Графики FACS (С) и гистограммы (D) показывают процентное и общее число вирус-специфических CD4 и CD8 Т-клеток в лёгких мышей линии BALB/c, получавших терапию крысиным Ig и антителами MC21.
E и F. Суспензии клеток медиастинальных лимфатических узлов приготовлены и исследованы на 18 час после инфицирования для на наличие дендритных клеток дыхательных путей, меченных CFSE. Показаны репрезентативные графики FACS (Е), а также процентные показатели и общее число клеток (F).
G и H. Процент апоптотических T-клеток субпопуляций CD4 и CD8 в легких, зараженных SARS-CoV, на 17 день. Показаны репрезентативные гистограммы (G) и процент апоптотических Т-клеток (H).
Данные репрезентативны для 2 независимых экспериментов (4–5 мышей/группа/эксперимент). Данные в пунктах (В), (D), (F) и (H).
представлены ± стандартная ошибка среднего (SEM).
*p < 0,05, **p < 0,01, и ***p < 0,001.
См. также дополнительный рисунок S5 в оригинале статьи.
Поскольку известно, что IFN-I сенсибилизирует Т-клетки к апоптозу (Carrero и соавт., 2004; Welsh и соавт., 2012), авторы измерили уровень апоптоза Т-клеток у зараженных мышей BALB/с и Ifnar⁻ᐟ⁻. Как показано на рис. 6G и 6H, более высокий процент апоптоза среди всех Т-клеток субпопуляций CD8 и CD4 был у инфицированных мышей линии BALB/c. Для исследования возможных механизмов Т-клеточного апоптоза был измерен уровень Fas и DR5 — рецепторов для Т-клеточных лигандов, индуцирующих апоптоз при связывании с FasL и TRAIL (Fujikura и соавт., 2013; Kayagaki и соавт., 1999). Как Fas на Т-клетках, так и его лиганд FasL на моноцитах, макрофагах и нейтрофилах, были повышены в группе BALB/c по сравнению с группой Ifnar⁻ᐟ⁻. Экспрессия TRAIL и DR5 была одинаковой в обеих группах мышей. Однако блокирование взаимодействия FasL-Fas или TRAIL-DR5 не снижало количество случаев апоптоза. Более того, авторы не наблюдали различий в экспрессии молекул, ассоциированных с внутренним путем апоптоза. Нейтрализация TNF, напротив, уменьшила количество случаев апоптоза Т-клеток, что указывает на прямую или косвенную роль TNF в этом процессе.
Обсуждение
Описанные здесь результаты показывают, что быстрая скорость репликации вируса SARS-CoV и относительная задержка в передаче сигнала от IFN-I способствовали накоплению воспалительных моноцитов и макрофагов,
повышению проницаемости сосудов и нарушению вирус-специфических Т-клеточных реакций, приводящих к тяжелому заболеванию мышей линии BALB/c, зараженных SARS-CoV. Эти результаты параллельно обнаруживаются у пациентов с SARS. У людей летальные исходы часто сопровождались начальной высокой вирусной нагрузкой (Chu и соавт., 2004) и иммунопатологией с повышенными уровнями IFN-I и цитокинов (Cameron и соавт., 2007). Увеличение количества макрофагов в легких было характерно для пациентов с тяжелой формой SARS (Franks и соавт., 2003; Nicholls и соавт., 2003). Экзогенный IFN-I, введенный до пика вирусного титра, блокирует этот каскад реакций и уменьшает иммунопатологию (рис. 2D и 2E).
Хотя противовирусные и иммуномодулирующие эффекты IFN-I хорошо известны в качестве защиты от вирусных инфекций, становится все более очевидным, что IFN-I также стимулирует неблагоприятные воспалительные реакции при некоторых бактериальных, грибковых и хронических вирусных инфекциях с помощью сложных механизмов, которые, как правило, являются патоген-специфическими (Carrero, 2013; McNab и соавт., 2015; Trinchieri, 2010). Таким образом, IFN-I способствует патологическим эффектам при внутриклеточных бактериальных инфекциях (возбудители M. tuberculosis, L. monocytogenes) путем подавления выработки ключевых молекул-эффекторов (TNF и IL-1) нейтрофилами и моноцитами, приводя к неконтролируемой репликации бактерий (Auerbuch и соавт., 2004; Mayer-Barber и соавт., 2014). При втором механизме IFN-I индуцирует фатальную иммунопатологию при некоторых грибковых инфекциях (Candida albicans) путем активации моноцитов и макрофагов, нейтрофил-опосредованной почечной иммунопатологии (Majer и соавт., 2012).
Кроме того, при персистирующих вирусных инфекциях, вызванных, например, вирусом лимфоцитарного хориоменингита или вирусом иммунодефицита обезьян (SIV), IFN-I способствует постоянной иммунной активации и увеличивает экспрессию ингибирующих молекул, таких как PD-1 и LAG-3, что приводит к нарушению Т-клеточных реакций (Sandler и соавт., 2014; Teijaro и соавт., 2013; Wang и соавт., 2012; Wilson и соавт., 2013). Отсутствие передачи сигнала от IFN-I при инфицировании штаммом PR8 вируса гриппа А (IAV) ведет к накоплению моноцитов с фенотипом Ly6Cint (со средней [intermediate] степенью экспрессии Ly6C), продуцирующих хемокин CXCL1. Последующая миграция нейтрофилов приводит к патологическим процессам в легких (Seo и соавт., 2011). Напротив, заражение IAV некоторых линий мышей привело к летальному исходу, и летальность снижалась при отсутствии передачи сигнала от IFN-I (Davidson и соавт., 2014). В отличие от тяжелых IAV-инфекций, отсутствие сигнала от IFN-I во время высоколетальной инфекции SARS-CoV не стимулировало нейтрофильную инфильтрацию легких, но значительно уменьшало накопление моноцитов и макрофагов (рис. 3 и S3A).
Инфекция вирусом SARS-CoV у мышей линии BALB/c характеризуется быстрой репликацией вируса, с пиком титров между 16 часом и вторым днем после заражения (рис. 2А). В то время как быстрая репликация вируса, вероятно, способствует неблагоприятным исходам, вирус-опосредованная задержка передачи сигнала от IFN-I и относительная резистентность SARS-CoV к IFN-I и его эффекторам, вероятно, играет роль. SARS-CoV абортивно (т. е. без образования новых вирионов) заражает макрофаги и дендритные клетки человека, которые минимально индуцируют IFN-I (Cheung и соавт., 2005; Law и соавт., 2005). Кроме того, SARS-CoV кодирует белки, которые ингибируют индукцию или передачу сигнала от IFN, а также функции эффекторных молекул (Totura и Baric, 2012).
Повышенный ответ IFN-I сопровождался выделением лиганда к рецептору хемокинов CCR2 (рис. 2С и S3B-S3D), что приводило к миграции моноцитов и макрофагов в очаг воспаления у мышей, инфицированных SARS-CoV. Это дополнительно увеличивало выделение IFN-I, лиганда CCL2 и других медиаторов воспаления (рис. 2 и S3C). Несмотря на то, что моноциты и макрофаги являются патогенетическим звеном у мышей линии BALB/c, зараженных SARS-CoV, эти клетки могут играть как защитную роль, так и быть патогенными, что зависит в первую очередь от самого патогена. В других моделях ТОРС у мышей с отсутствием транскрипционного фактора STAT1, зараженных SARS-CoV, макрофаги были патогенными, но и в этом случае патологические изменения в легких вызваны преобразованием фенотипа макрофагов из M1 (классического) в M2 (альтернативно активированного), что приводило к увеличению фиброза и неблагоприятных исходов. У этих мышей блокирование передачи сигнала от STAT-1 в субпопуляциях моноцитов и макрофагов способствовало развитию легочной патологии (Frieman и соавт., 2010; Page и соавт., 2012; Zornetzer и соавт., 2010). При респираторно-синцитиальном вирусе (RSV), вирусе простого герпеса (HSV) и вирусе Западного Нила (WNV) моноциты и макрофаги элиминируют вирусы, являясь защитными клетками (Goritzka и соавт., 2015; Terry и соавт., 2012). Напротив, у мышей, инфицированных штаммом PR8 IAV, данные клетки оказывают двойной эффект: способствуют развитию патологии легких в той же степени, в которой являются необходимыми для оптимальной вирус-специфической Т-клеточной реакции (Aldridge и соавт., 2009; Lin и соавт., 2008).
Несмотря на массивное скопление моноцитов и макрофагов в легких, пораженных SARS-CoV, механизмы, которые вызывают летальный исход остаются не ясными. Наши исследования показывают, что TNF является важным фактором в развитии летального исхода, так как его нейтрализация повышает выживаемость (рис. 4F), но вполне вероятно, что и другие цитокины, вырабатываемые моноцитами и макрофагами, такие как IL-6, IL-1β, NO-синтаза, среди других, оказывают неблагоприятные эффекты.
IFN-индуцированные провоспалительные цитокины запускают экспрессию TRAIL-DR5 и FasL-Fas на миелоидных клетках, а также Т-клетках, которые участвуют в Т-клеточном апоптозе (Boonnak и соавт., 2014; Daigneault и соавт., 2012; Kayagaki и соавт., 1999). Авторы выявили увеличение случаев апоптоза Т-клеток у линии BALB/c в сравнении с мышами Ifnar⁻ᐟ⁻, зараженными SARS-CoV, но в отличие от более ранних исследований (Boonnak и соавт., 2014), они обнаружили, что ни FasL-Fas, ни TRAIL-DR5, ни внутренние пути апоптоза не были вовлечены в этот процесс. Усилению Т-клеточного апоптоза, вероятно, способствовал субоптимальный Т-клеточный ответ (рис. 6). Т-клетки сдерживают цитокиновый шторм (Kim и соавт., 2007), подавляя ответ системы врожденного иммунитета. Поэтому субоптимальные Т-клеточные реакции могут привести к неконтролируемому ответу от клеток врожденного иммунитета с длительным выделением провоспалительных медиаторов и последующей иммунопатологией.
Подводя итог, авторы демонстрируют, что отсроченная экспрессия IFN-I вызывает неадекватную воспалительную реакцию с последующими иммунопатологическими процессами в легких при инфекции, вызываемой SARS-CoV. Более того, минимальные различия в вирусной нагрузке в легких у мышей BALB/c и Ifnar⁻ᐟ⁻ и полная защита от развития заболевания после деплеции моноцитов и макрофагов предполагают, что IFN-I-обусловленные иммунопатологические изменения приводят к увеличению заболеваемости и летальности от SARS-CoV, вне зависимости от степени репликации вируса. Защитный эффект от раннего, но не позднего введения IFN-I (рис. 2D; Haagmans и соавт., 2004; Kumaki и соавт., 2011) и патологические эффекты отсроченного IFN-I-опосредованного ответа позволяют предположить, что IFN-I следует с осторожностью применять при лечении зараженных пациентов. В будущем вместе со снижением начальной вирусной нагрузки с помощью противовирусных препаратов, терапевтические подходы, которые уменьшают иммунопатологические изменения, могут помочь снизить высокие показатели летальности, ассоциированные с возникающими коронавирусными и, возможно, другими высокопатогенными вирусными заболеваниями.
