
Неонатальный скрининг

Неонатальный скрининг, ласково именуемый в нашей стране «пяточка», является одним из первых важных исследований новорожденного. В России скрининг преимущественно направлен на выявление пяти наследственных болезней обмена: фенилкетонурии, врожденного гипотиреоза, врожденной дисфункции коры надпочечников (ВДКН), галактоземии и муковисцидоза. За рубежом этот список расширен до 50 различных заболеваний, в некоторых штатах Америки их свыше 60. Здоровый доношенный новорожденный допускается к скринингу на 4–5 сутки, недоношенный — на седьмой день после рождения. Заболевания, на выявление которых направлен скрининг, никак не проявляют себя в периоде новорожденности, но их ранняя диагностика и своевременно начатое патогенетическое лечение существенно влияют на прогноз и качество жизни ребенка. Помимо исследования крови проводится аудиометрия для оценки слуха и пульсоксиметрия для скрининга пороков сердца, но в данной статье мы преимущественно сосредоточимся на тестировании крови.
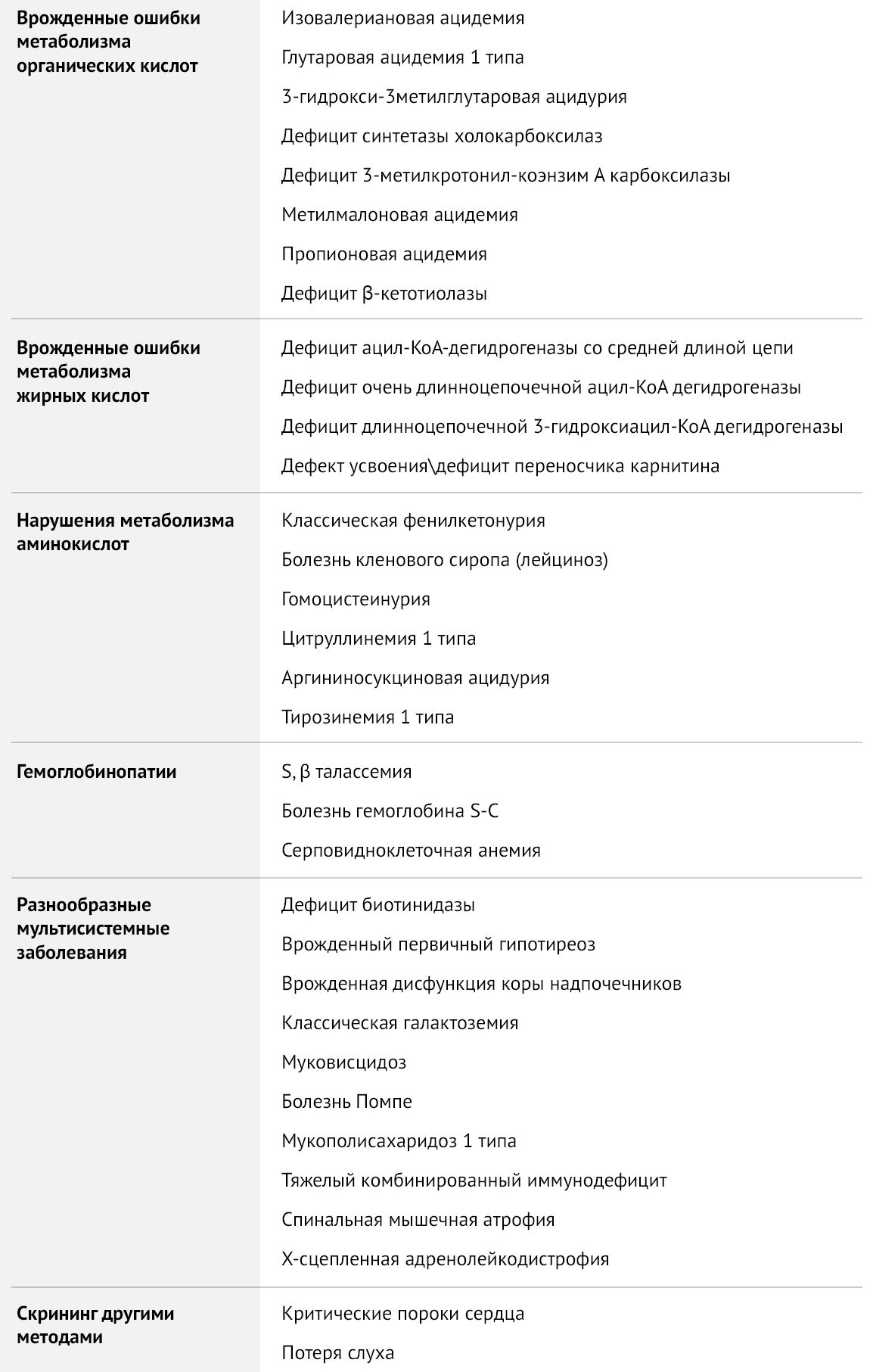
Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
Обязательный скрининг
На 4–5 сутки после рождения здорового доношенного ребенка или на седьмые сутки жизни недоношенного ребенка проводится тестирование методом «сухого пятна».
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.
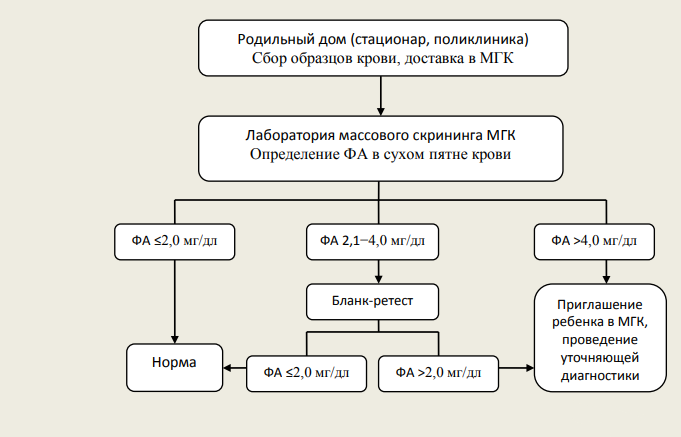
Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.

Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Расширенный скрининг
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Аминоацидопатии
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Аргининосукциновая ацидурия вызывается мутациями в гене ASL, который кодирует фермент аргининосукцинатлиазу. Этот фермент катализирует превращение аргинино-янтарной кислоты в аргинин и фумарат на четвертом этапе цикла мочевины. Дефекты на этой стадии цикла мочевины приводят к накоплению в плазме аммиака, аргинино-янтарной кислоты, цитруллина и оротовой кислоты в моче, а также к дефициту аргинина в плазме. Ацидурия может иметь различную клиническую картину с началом в любом возрасте, включая период новорожденности. Состояние новорожденных обычно не вызывает подозрений в течение первых 24–48 часов после рождения, но в течение нескольких дней дебютирует тяжелая гипераммониемия, проявляющаяся летаргией, сонливостью, отказом от еды, рвотой, тахипноэ и респираторным алкалозом. Если не начать лечение, может произойти обострение летаргии, судороги, кома и смерть. Позднее начало ацидурии обычно индуцировано острой инфекцией, стрессом или высоким потреблением белка. Сообщалось также о поздних когнитивных дефектах или нарушениях обучаемости при отсутствии эпизодов гипераммониемии. У некоторых пациентов заболевание может протекать бессимптомно, несмотря на четкие биохимические признаки.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.
Источники:
- Rink B., Dukhovny S. Newborn Screening and the Role of the Obstetrician-Gynecologist //OBSTETRICS AND GYNECOLOGY. – 2019.
- Merritt J. L., II M. N., Kanungo S. Fatty acid oxidation disorders //Annals of translational medicine. – 2018.
- Еремина Е. Р. Клинический случай редкой органической ацидурии //Медицинская генетика. – 2018.
- Cherella C. E., Wassner A. J. Congenital hypothyroidism: insights into pathogenesis and treatment //International journal of pediatric endocrinology. – 2017.
- Чикулаева О. А. Федеральные клинические рекомендации по диагностике и лечению врожденного гипотиреоза у детей //Проблемы эндокринологии. – 2014.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017.
- Клинические рекомендации “Федеральные клинические рекомендации по оказанию медицинской помощи детям с галактоземией”, 2015
- Клинические рекомендации “Кистозный фиброз (муковисцидоз)”, 2020
- Клинические рекомендации “Гомоцистеинурия”, 2016
- Клинические рекомендации “Клинические рекомендации по ведению и терапии новорожденных с заболеваниями надпочечников”. 2016
- De Laet C. et al. Recommendations for the management of tyrosinaemia type 1 //Orphanet journal of rare diseases. – 2013.
- Nagamani S. C. S., Erez A., Lee B. Argininosuccinate lyase deficiency //Genetics in medicine. – 2019.
