
Ключевые особенности опухолевого метаболизма

Введение
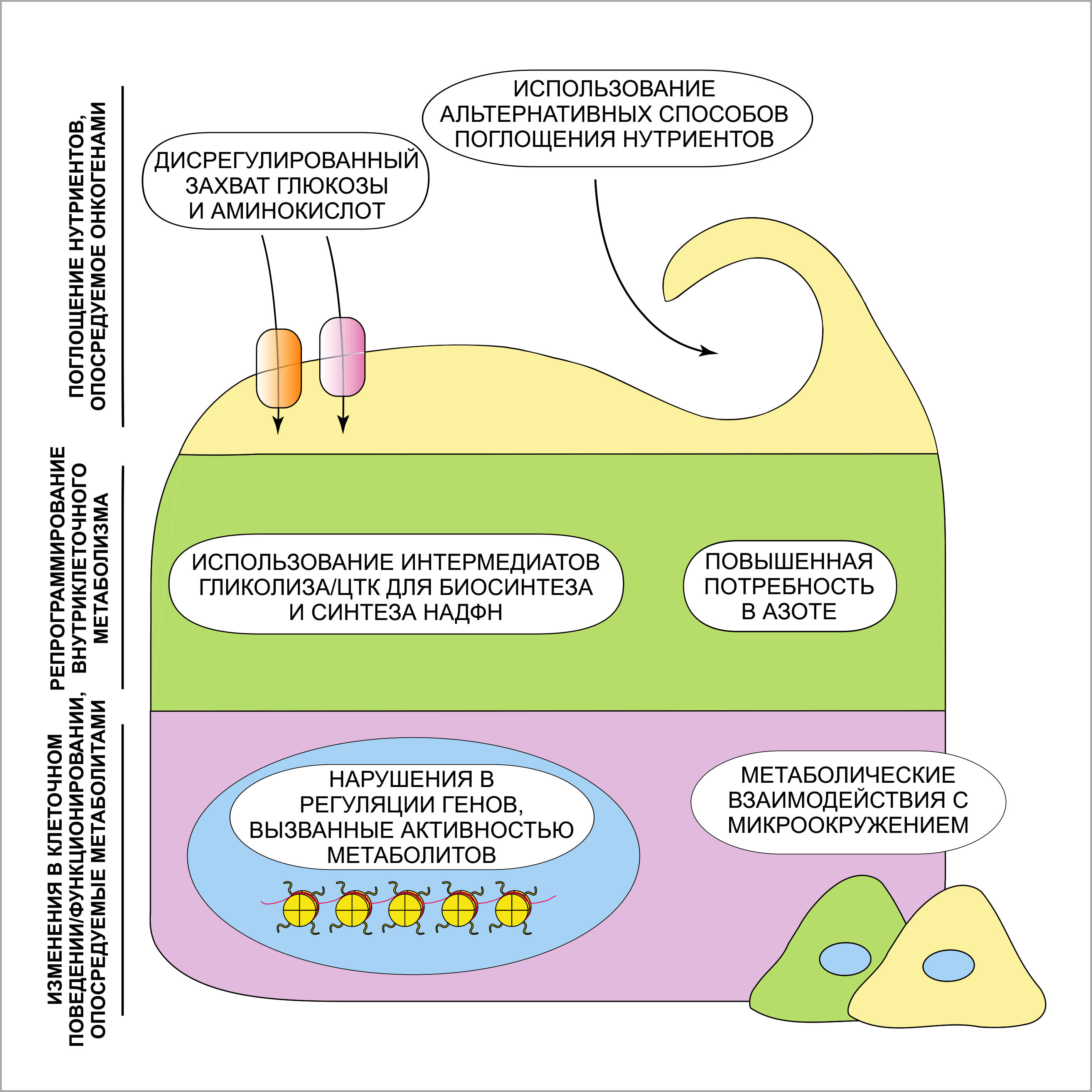
Первые наблюдения по метаболическим нарушениям, характерным для неоплазий, были сделаны около века назад, а в течение последнего десятилетия интерес к опухолевому метаболизму резко возобновился. Благодаря новым биохимическим и молекулярно-биологическим методикам, исследования обмена веществ опухолевых клеток расширили наше понимание механизмов и результатов опухоль-ассоциированных метаболических нарушений на различных этапах опухолеобразования. В частности, стало ясно, что такие нарушения затрагивают все этапы взаимодействий клетки с продуктами обмена, в числе которых: (1) влияние на поступление метаболитов посредством повышения способности к захвату необходимых питательных веществ, (2) определение порядка вовлечения нутриентов в метаболические пути согласно их опухолегенным свойствам и (3) оказание долгосрочного эффекта на клеточное развитие, в числе которых нарушения в дифференцировке опухолевых клеток и компонентов их микроокружения (Рисунок 1). В данном обзоре особое внимание уделяется различным ключевым признакам ассоциированного с опухолеобразованием перепрограммирования метаболизма, а также влиянию этих признаков на установление и сохранение опухолегенного состояния.
Неадекватное поглощение глюкозы и аминокислот
Для того чтобы восполнить затраты на биосинтетические потребности, ассоциированные с пролиферацией, клетке необходимо наращивать импорт питательных веществ извне. Двумя наиболее значимыми нутриентами, обеспечивающими выживание и биосинтетические процессы в клетках млекопитающих, являются глюкоза и глутамин. Катаболизируя эти молекулы, клетка получает группы различных углеродсодержащих продуктов обмена, которые используются в качестве строительных блоков при сборке широкого спектра макромолекул. Кроме того, контролируемое окисление углеродных скелетов глюкозы и глутамина позволяет клетке получать восстановленную энергию в форме НAДH или ФAДH2, опосредующих перенос электронов в электрон-транспортной цепи для синтеза АТФ, или же в форме НAДФH, участвующего во многих процессах биосинтеза, а также способствующего сохранению окислительно-восстановительного потенциала.
Заметно повышенное потребление глюкозы в опухолях по сравнению с непролиферирующими нормальными тканями впервые было описано более 90 лет назад немецким физиологом Отто Варбургом. Это наблюдение было подтверждено при исследованиях многих опухолей, также была установлена корреляция данного явления с неблагоприятным для онкологических пациентов прогнозом. Визуализация захвата меченого радиоактивным фтором аналога глюкозы, 18F-фтордезоксиглюкозы (18F-ФДГ) на основе позитронной эмиссионной томографии (ПЭТ) была успешно испытана в клинике для диагностики и установления стадии развития опухоли, а также контроля за ответной реакцией организма на лечение.
Глутамин, второй по важности субстрат, способствующий росту, требует для биосинтеза de novo различных азотсодержащих соединений не только углерода, но и азота в восстановленной форме. Таким образом, глутамин поставляет азот, необходимый для биосинтеза пуриновых и пиримидиновых нуклеотидов, глюкозамин-6-фосфата, а также заменимых аминокислот. Однако, если заменимые аминокислоты могут образовываться в клетках млекопитающих de novo, то незаменимые должны поступать извне. Примечательно, что импорт незаменимой аминокислоты лейцина через заякоренный в плазматической мембране антипорт нейтральных аминокислот LAT1 сопряжён с единовременным выбросом глутамина из клетки. Таким образом, внутриклеточный глутамин может также способствовать импорту широкого диапазона субстратов LAT1, включая лейцин, изолейцин, валин, метионин, триптофан и фенилаланин.
Повышенную потребность пролиферирующих опухолевых клеток в глутамине впервые отметил в 1950-х гг. американский физиолог Гарри Иггл, продемонстрировавший, что для оптимального роста культуры клеток HeLa необходим 10-100-кратный молярный избыток глутамина в среде относительно других аминокислот. Более того, было установлено, что глутамин – наиболее интенсивно потребляемая аминокислота в асцитных карциномах Эрлиха, а также в некоторых гепатомах и карциносаркомах, пролиферирующих in vivo. В действительности, значительное множество опухолегенных очагов характеризуются опустошением запасов глутамина из опухолевого микроокружения, в сравнении с аналогичной нормальной тканью. Успехи визуализации благодаря 18F-ФДГ стали предпосылкой для недавних исследований, продемонстрировавших потенциал 18F-маркированных глутаминовых меток в доклинических и ранних клинических испытаниях. Использование 18F-маркированного глутамина в качестве метки позволяет собирать потенциально полезную информацию об опухоли в тех случаях, когда применение 18F-фтордезоксиглюкозы нецелесообразно, например, при визуализации опухолей, локализованных в органах с повышенным потреблением глюкозы, таких как мозг.
Что заставляет опухолевые клетки поглощать глюкозу и глутамин в больших количествах? В большинстве случаев, вопреки наличию богатых питательными веществами плазмы и внеклеточной жидкости вокруг, клетки многоклеточных эукариот не импортируют нутриенты постоянно. С другой стороны, захват питательных элементов напрямую регулируется сигнальными путями факторов роста (Рисунок 2). К примеру, в отсутствие факторов роста гематопоэтические и нейрональные клетки не могут поглощать глюкозу в количествах, необходимых для поддержания клеточной биоэнергетики. Это, в свою очередь, оказывает отрицательное влияние на размеры клетки, митохондриальную активность и образование АТФ – и всё это несмотря на избыток глюкозы в культуральной среде. Однако жизнеспособность лишённых факторов роста клеток может быть восстановлена благодаря единовременной экспрессии транспортера глюкозы GLUT1 на клеточной мембране и первого фермента гликолитического пути – гексокиназы (ГК).
Вдобавок к растворимым факторам роста, взаимодействия клеток с внеклеточным матриксом играют важную роль в регулировании захвата глюкозы. Культивирование эпителиоцитов молочной железы в условиях, при которых клетки отделены от внеклеточного матрикса, нарушает захват глюкозы и приводит к снижению митохондриальной активности и уровня образования АТФ. В целом, эти наблюдения показывают, что вход глюкозы в клетки не обеспечивается её сиюминутными биоэнергетическими потребностями, но напрямую зависит от воздействия внеклеточных стимулов.
В сравнении со здоровыми клетками-эквивалентами, которым для выживания и пролиферации необходимы механизмы сигнальной трансдукции, опосредованной адгезией и факторами роста, опухолевые клетки аккумулируют в себе онкогенные повреждения, помогающие им обрести значительную независимость от этих экзогенных элементов. В частности, генетические нарушения, поражающие PI3-киназу, её негативные регуляторы PTEN и INPP4B, а также активирующие мутации и амплификации генов в различных тирозинкиназных рецепторах, ведут к конститутивному (непрерывному) захвату глюкозы и её метаболизированию во многих типах опухолей.
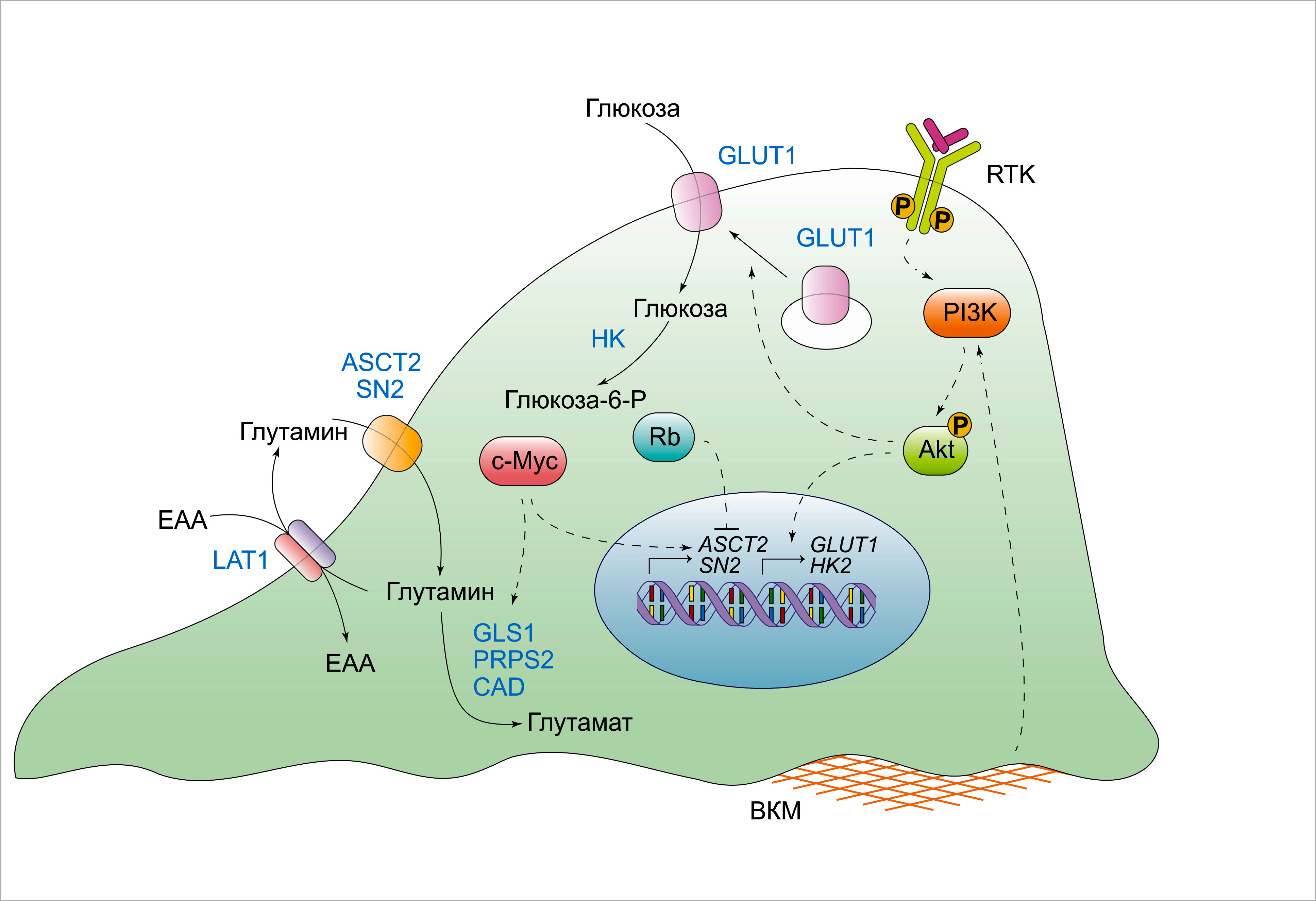
RTK, тирозинкиназный рецептор; GLUT1, переносчик глюкозы 1; ASCT2/SN2, переносчик глутамина; LAT1, переносчик нейтральных аминокислот; EAA, незаменимые аминокислоты; GLS1, глутаминаза 1; PRPS2, фосфорибозилпирофосфат-синтетаза 2; CAD, карбамоилфосфат-синтетаза 2; НК, гексокиназа; ECM, внеклеточный матрикс.
Точка конвергенции сигналов от рецептора тирозинкиназ и от внеклеточного матрикса – сигнальный путь PI3K/Akt – играет важнейшую роль в регуляции захвата глюкозы. Этот сигналинг одновременно усиливает как экспрессию мРНК транспортера глюкозы GLUT1, так и перемещение белка GLUT1 с мембран внутренних компартментов на поверхность клетки. Кроме этого, Akt повышает активность ГК, фосфорилирующей молекулы глюкозы, что предупреждает их выход наружу во внеклеточное пространство, а также фермента фосфофруктокиназы (ФФК), катализирующего ключевую необратимую стадию гликолиза. В действительности, интенсивность сигнала 18F-ФДГ-ПЭТ в опухолях плотно коррелирует с уровнем активности PI3K/Akt-пути; она ослабляется PI3-киназой и ингибиторами рецептора тирозинкиназ. Более того, экзогенная экспрессия лишь только конститутивно активной формы Akt сама по себе может стимулировать гликолиз, причём этого количества достаточно для восстановления размеров, жизнеспособности, митохондриальной активности и уровня АТФ в клетках, лишённых факторов роста, что зависит только от наличия глюкозы в культуральной среде. Конститутивно активная форма Akt также предупреждает снижение уровня АТФ вследствие потери клеточной адгезии. Помимо этого, сигнальный путь Akt играет ключевую роль в поглощении глюкозы при условиях повышенного уровня биосинтетических процессов. К примеру, таргетная делеция Akt1 в молочной железе мыши устраняет индуцированное лактацией повышение захвата молекул глюкозы, что ведёт к недостаточному образованию молока.
Тем не менее, PI3/Akt-сигнальный путь – не единственный онкогенный стимул, принимающий участие в захвате глюкозы. Было показано, что другие онкогенные сигнальные белки – к примеру, Ras – повышают экспрессию мРНК гена GLUT1 и увеличивают объёмы потребления глюкозы клеткой. Таким образом, различные узлы сигнальных путей факторов роста, ошибочно активированные в опухолях, делят между собой влияние на содействие клеточному поглощению глюкозы – основного метаболического субстрата.
Пути сигнальной трансдукции, регулирующие поглощение глутамина, изучены не полностью. Транскрипция фактора c-myc, зачастую амплифицированного в различных опухолях, особенно выражена в быстро пролиферирующих клетках; он играет важнейшую роль в процессе утилизации глутамина делящимися клетками. Кроме этого, c-myc индуцирует транскрипцию транспортеров глутамина ASCT2 и SN2 и повышает экспрессию глутамин-метаболизирующих ферментов: глутаминазы (GLS1), фосфорибозилпирофосфатсинтетазы (PRPS2) и карбамоилфосфатсинтетазы-2 (в составе трифункционального белка CAD), которые поддерживают опосредованное транспортерами поглощение глутамина посредством его конвертации в глутамат. Образующийся глутамат не имеет возможности покинуть клетку через транспортер глутамина; по мере накопления, он активизирует цикл Кребса и стимулирует захват цистеина, выступая в роли обменного субстрата для антипортера цистеина xCТ.
Вдобавок к положительному регулирующему эффекту фактора c-myc, на поглощение глутамина действует также и негативная регуляция – со стороны семейства белковых опухолевых супрессоров Rb. Было показано, что «выключение» из работы белков из этого семейства ведёт к увеличению потребления и утилизации глутамина посредством E2F-зависимого повышения экспрессии ASCT2 и GLS1. Факторы c-myc и E2F – это два ключевых координатора клеточного деления; их эффект проявляется, в частности, в предоставлении клеткам доступа к глутамину – метаболическому субстрату, незаменимому в процессах биосинтеза, которые сопровождают репликацию ДНК.
Для любой отдельно взятой клетки попытка к пролиферации в отсутствии нормального обеспечения ресурсами может обернуться катастрофическими последствиями. Для того чтобы их избежать, активированные по ошибке онкогены и/или потеря опухолевых супрессоров задерживают опухолевые клетки в состоянии, при котором они конститутивно поглощают доступные для них вещества – глюкозу, глутамин, незаменимые аминокислоты – из внеклеточного микроокружения, что, в свою очередь, ведёт к их неконтролируемой пролиферации.
Использование альтернативных путей для получения питательных веществ
Несмотря на высокую интенсивность поглощения глюкозы и аминокислот, опухолевые клетки in vivo зачастую сталкиваются с условиями нехватки нутриентов вследствие усиленного их расходования и неадекватного снабжения ими со стороны опухолевой сосудистой сети. В целях борьбы с нарушенной системой доставки нормальных анаболических субстратов, некоторые опухоли приобрели мутации, активирующие клеточную способность к использованию альтернативных путей получения необходимых нутриентов. К примеру, плазматическая и межклеточная жидкости богаты растворёнными белками, однако внеклеточные белки обычно не используются клеткой в качестве источника аминокислот. Примечательно, что экспрессия мутантных аллелей Ras или c-Src обеспечивает клетки механизмом извлечения свободных аминокислот из продуктов лизосомальной деградации внеклеточных белков (Рисунок 3A). По сравнению с поглощением низкомолекулярных питательных веществ, поступающих в клетку посредством специализированных транспортеров, захват внеклеточных макромолекул протекает благодаря макропиноцитозу – процессу, при котором из частей внеклеточной жидкости формируются крупные везикулы. Макропиноцитоз стимулируется перестройкой актинового цитоскелета, опосредованного Ras и c-Src. Наполненные жидкостью макропиносомы переходят во внутреннюю среду клетки, где они сливаются с лизосомами, и поглощённые белки подвергаются протеолитической деградации, в результате чего образуются свободные аминокислоты. Так, обогащение культуральной среды сывороточным альбумином в физиологической концентрации (20-30 мг/мл) активирует пролиферацию KRASG12D-трансформированных эмбриональных фибробластов мыши в отсутствие незаменимой аминокислоты лейцина. Было также установлено, что восполняющее введение альбумина способствует пролиферации опосредованной мутацией KRASG12D опухолевой линии поджелудочной железы мыши в культуральной среде, лишённой всех аминокислот в свободном состоянии. В отличие от эффекта мутации KRASG12D, сигнальный путь PI3K/Akt не усиливает потребление внеклеточного белка клеткой, несмотря на способность этого пути стимулировать поглощение низкомолекулярных нутриентов.
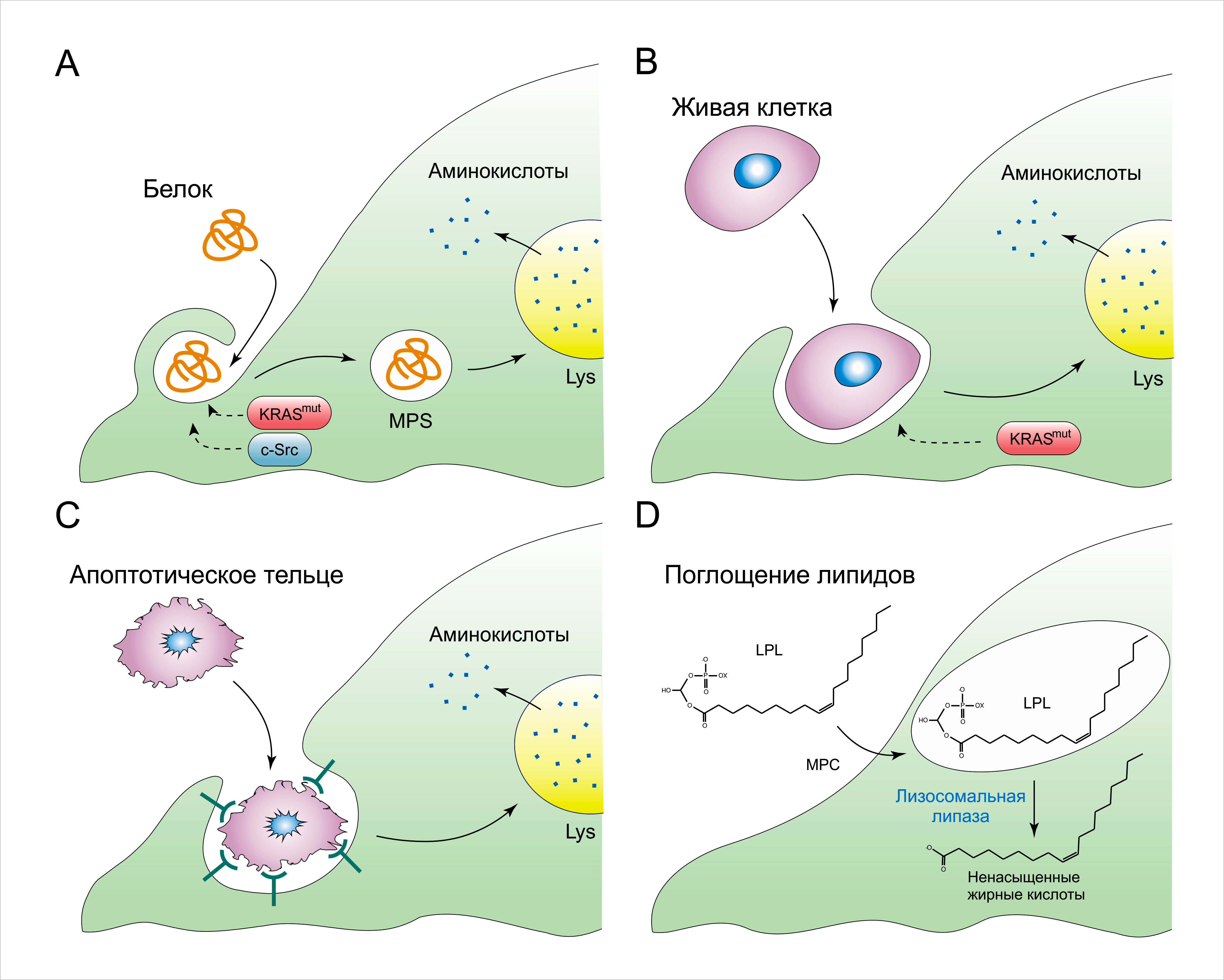
Примечательно, что ингибиторы mTORC1 сильно повышают клеточную способность к получению аминокислот из поглощённых внеклеточных белков и усиливают их рост в отсутствии незаменимых аминокислот. Таким образом, утилизация внеклеточных белков в качестве нутриентов ингибируется комплексом mTORC1 в условиях дефицита аминокислот и используется лишь как экстренный ресурс в ситуации, при которой свободные аминокислоты недоступны. Пространственное отличие эффекта от лечения рапамицином мышей с мутацией KRASG12D в модели опухоли поджелудочной железы даёт хорошую иллюстрацию для нашей парадигмы. Наряду с тем, что рапамицин подавляет пролиферацию опухолевых клеток в краевых участках опухоли, где сосудистое снабжение питательными веществами достаточное, это вещество также усиливает клеточную пролиферацию в более глубоких плохо васкуляризированных слоях опухоли благодаря стимуляции лизосомальной деградации внеклеточных белков.
Помимо растворённых белков плазмы, свободные аминокислоты могут быть получены путём поглощения и переваривания целых живых клеток через процесс, известный как энтоз (Схема 3В), а также через фагоцитоз апоптотических клеточных телец (Рисунок 3С). Аналогичным образом, частицы, поглощённые посредством энтоза или фагоцитоза, расщепляются в лизосомах, что обеспечивает достаточное снабжение клетки для выживания и пролиферации в условиях аминокислотного дефицита. Интересно отметить, что клетки, имеющие мутантный аллель KRAS, с большей вероятностью осуществят энтоз, чем сами будут вовлечены в процесс поглощения. Таким образом, мутантные клетки с KRAS не только имеют преимущество в плане обеспечения нутриентами по сравнению с их немутированными соседями, но и активно элиминируют последних. Такое свойство может инициировать межклеточную конкуренцию внутри опухоли, в результате чего появятся более агрессивные клеточные популяции.
Другим результатом отсутствия сосудистого обеспечения клеток питательными веществами является появление ишемизированных тканей, испытывающих гипоксию, что ведёт к подавлению многих реакций биосинтеза, для которых в качестве акцептора электронов необходим кислород. Например, гипоксия влияет на появление двойных связей, катализируемых стеарол-СоА-десатуразой (SCD1) в de novo синтезированных жирных кислотах, что ведёт к дефициту ненасыщенных жирных кислот. Для того чтобы восполнить потерянные жирные кислоты, находящиеся в условиях гипоксии, клетки импортируют «уже готовые» ненасыщенные жирные кислоты из окружающей среды в форме лизофосфолипид с одним ацильным хвостом (Рисунок 3D). В соответствии с этим, исчезновение липидов сыворотки ведёт к ER-стрессу и апоптозу некоторых опухолевых клеток, культивированных в условиях гипоксии.
В других опухолевых клетках наблюдается усиленное выделение во внеклеточное пространство свободных жирных кислот, образованных из более сложных липидов. Повышенная экспрессия липопротеин-липазы и моноацилглицерол-липазы (MAGL), обнаруженная на примере многих неоплазий, коррелирует с инвазионной способностью новообразований. Наконец, в то время как одни опухоли вырабатывают адаптации по поглощению жирных кислот напрямую из плазмы, другие стимулируют выброс запасённых липидов из нормальных соседних клеток. К примеру, появление белка FABP4, связывающего длинноцепочечные жирные кислоты, на поверхности метастазирующих опухолевых клеток яичника помогает им захватывать эти молекулы напрямую из адипоцитов сальника.
Таким образом, опухолевые клетки могут эксплуатировать различные пути для получения питательных веществ, способствующих их выживанию и пролиферации в метаболически неблагоприятных условиях. Эти адаптации обуславливают способность к поглощению недоступных при обычных условиях нутриентов, а также промежуточных продуктов обмена, если их клеточный синтез нарушен.
Было выявлено, что даже в отсутствие нутриентов во внеклеточной среде опухолевые клетки могут перестраивать метаболические пути для поддержания своей жизнеспособности. В частности, благодаря катаболическому процессу макроаутофагии они достаточно длительно могут выдерживать периоды нехватки питательных веществ в среде. Макроаутофагия характеризуется обволакиванием внутриклеточных макромолекул и целых органелл двумембранными структурами и их слиянием с лизосомами. Однажды попав в лизосому, содержимое незамедлительно подвергается деградации резидентными протеазами и липазами, в результате чего высвобождаются свободные амино- и жирные кислоты. Аутофагия способствует сохранению жизнеспособности клеток в условиях отсутствия нутриентов или факторов роста в культуре в течение недель. Кроме этого, было выявлено, что делеция важнейшего компонента механизма аутофагии, Atg7, ведёт к резкому переходу опухолей лёгкого (инициированных онкогенами KrasG12D и BrafV600E) из злокачественной аденокарциномы в доброкачественную онкоцитому. Тем не менее, аутофагия не может обеспечить клетки новой биомассой и, следовательно, не способна поддерживать пролиферацию в условиях нехватки питательных веществ.
Роль гликолиза/цикла Кребса в биосинтезе и образовании НАДФН
Клеточная пролиферация не только повышает запросы клетки по объёму необходимых ей питательных веществ, как это обозначено выше, но также меняет способы их утилизации. В этой связи, в то время как опухолевая клетка неактивна, глюкоза идёт преимущественно на образование ацетил-КоА в митохондриях, который далее вступает в окислительные процессы в цикле трикарбоновых кислот (ЦТК, цикл Кребса). Электроны, полученные от реакций окисления в ЦТК, переносятся посредством НАД+/НАДН или ФАД/ФАДН2 к электронной транспортной цепи, благодаря чему создаётся электрохимический градиент для синтеза АТФ.
Использование углеродных молекул быстро пролиферирующими клетками и теми, что находятся в покоящемся состоянии, сильно отличается друг от друга (Рисунок 4A). Главный способ использования дефицитных углеродных соединений в интенсивно делящихся клетках заключается в биосинтезе различных биомолекул, среди которых жирные кислоты и холестерол, производные пентозных и гексозных сахаров, глицерол, нуклеотиды и заменимые аминокислоты. Для осуществления этого, быстро пролиферирующая клетка должна для начала превратить полученные нутриенты в различные пулы промежуточных метаболитов (Рис. 4В). Эти молекулы включают: цитозольные ацетил-КоА, переносящие одноуглеродные группы производные фолиевой кислоты, S-аденозилметионин (SAM), а также некоторые интермедиаты гликолиза и ЦТК. Кроме того, многие реакции биосинтеза по своей природе носят восстановительный характер, и поэтому нуждаются в соответствующем источнике энергии. К примеру, для образования пальмитиновой кислоты необходимо 14 восстановительных эквивалентов, тогда как для постройки молекулы холестерола нужно 26. Донором восстановительных эквивалентов для клеточных биосинтетических реакций служит НАДФН. Образование НАДФН из НАДФ+ достигается благодаря окислению углеродных субстратов в метаболических путях, отличных от тех, что поставляют НАДН для поддержки митохондриального транспорта электронов. Таким образом, для активно делящихся клеток важно распределить часть углеродных субстратов на синтез НАДФН.
Репрограммирование углеродного метаболизма интенсивно делящимися клетками в какой-то мере объясняет оригинальные наблюдения Варбурга на тему обмена веществ в опухолях. Экспериментируя с инкубированными с глюкозой ex vivo кусочками опухоли, Варбург показал, что несмотря на поглощение глюкозы в огромных количествах и наличие доступа к кислороду, клетки опухоли не получили преимуществ от сопряжения гликолиза с ЦТК, несмотря на биоэнергетическую выгоду такого явления. Напротив, выращенные в богатой глюкозой среде клетки конвертировали избыток пирувата в лактат, который, в свою очередь, отправлялся во внеклеточное пространство. Принимая за условие то, что большая часть глюкозы в клетках идёт на образование АТФ, Варбург и многие другие исследователи неверно интерпретировали данный феномен, объясняя его «необратимым повреждением клеточного дыхания», что оставляет опухолевые клетки со значительно менее эффективным путём образования АТФ посредством реакций субстратного фосфорилирования и гликолиза.
Однако последующие исследования на тему состояния митохондрий в опухолевых клетках показал, что, в общем и целом, эти клетки сохраняют митохондрии в функциональном состоянии, способном осуществлять окислительное фосфорилирование. Интересно, что направленное нарушение структуры митохондриальной ДНК снижает неопластический потенциал опухолевых клеточных линий in vitro и in vivo. Более того, преимущественная конверсия глюкозы в лактат была описана в генетически нормальных делящихся клетках, а также в клетках, инфицированных вирусами. Эти наблюдения предполагают, что эффект Варбурга является, скорее, регулируемым метаболическим состоянием, нежели адаптацией из-за дефектного клеточного дыхания и может, по видимости, быть полезным в период повышенных биосинтетических потребностей.
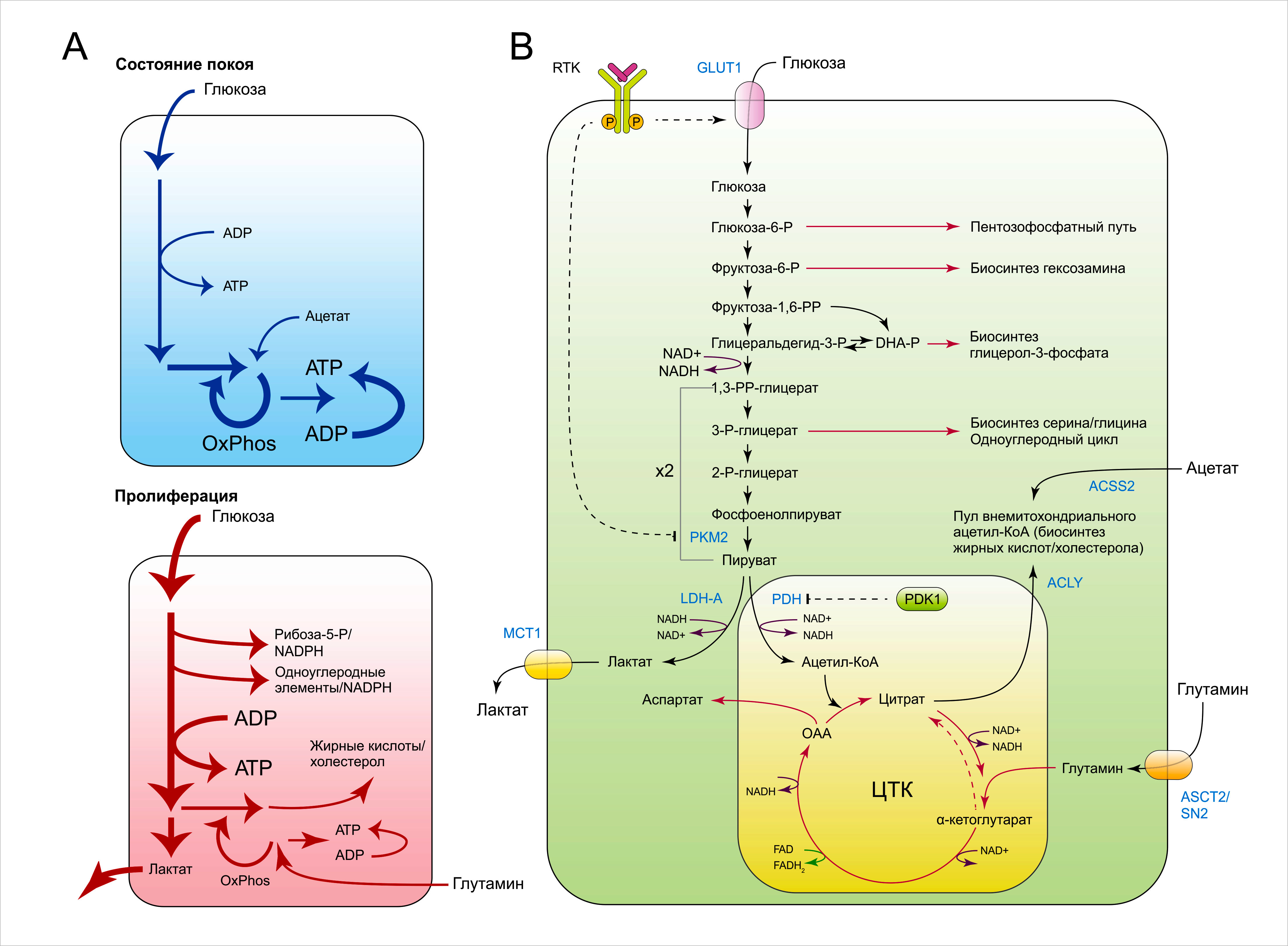
RTK, тирозинкиназный рецептор; GLUT1, переносчик глюкозы 1; PKM2, пируваткиназа M2; ACSS2, aцетил-CoA-синтетаза 2; LDH-A, лактатдегидрогеназа A; PDH, пируватдегидрогеназа; PDK1, пируватдегидрогеназная киназа 1; ACLY, ATФ-цитратлиаза; MCT1, монокарбоксилатный переносчик 1; ASCT2/SN2, переносчик глутамина.
Тогда почему быстро пролиферирующие клетки превращают избыток пирувата в лактат вместо того, чтобы переносить этот избыток в митохондрии для осуществления окислительного фосфорилирования? Только теперь ответ начинает всплывать на поверхность. Как ни странно, в этих клетках наблюдается лишь умеренный прирост потребления АТФ относительно их нужд, касающихся исходных для постройки полимеров молекул и восстановительных эквивалентов в виде НАДФН. Катаболизм глюкозы играет важную роль в обеспечении клетки этими молекулами-предшественниками и восстановительными эквивалентами. С другой стороны, активность ЦТК, благодаря которому производятся НАДН и АТФ, является главным негативным регулятором метаболизма глюкозы. Конвертируя избыток пирувата в лактат, активно делящиеся клетки предотвращают накопление НАДН в цитозоле и тем самым снижают образование АТФ, что усиливает метаболизирование глюкозы в цитоплазме. Таким образом, эта система уже становится свободной от репрессии по типу обратной связи со стороны избытка АТФ, образованного в митохондриях.
Понимание этого стало отправной точкой для исследований всего того, что может дать быстро пролиферирующим клеткам разобщение процессов гликолиза и окислительного фосфорилирования. Согласно классическому описанию, гликолиз представляет собой единую цепь молекулярных событий, ведущих к образованию пирувата; многие промежуточные метаболиты гликолиза могут вступать в разветвлённые метаболические пути, в результате чего образуются различные биосинтетические молекулы-предшественники. Важнейшим из этих разветвлённых путей является пентозофосфатный путь (ПФП), в котором глюкоза-6-фосфат частично окисляется с образованием НАДФН и рибоза-5-фосфата — структурного компонента нуклеотидов. Активность ПФП зачастую повышается при опухолеобразовании. Экспрессия ключевых ферментов этого пути, транскетолазо-подобного белка 1 (transketolase-like 1, TKTL1) и трансальдолазы (transaldolase, TALDO), часто повышена в опухолях. Было продемонстрировано, что как онкогены, так и онкосупрессоры участвуют в регуляции активности ПФП. К примеру, Ras-опосредованная трансформация вызывает усиление транскрипционной активности ферментов, принимающих участие в биосинтезе рибоза-5-фосфата. Более того, белок р53 дикого типа (но не мутантный) подавляет ПФП, напрямую связывая и инактивируя лимитирующий скорость ПФП фермент глюкоза-6-фосфат-дегидрогеназу (Г6ФД).
Фруктоза-6-фосфат, следующий продукт гликолитического пути после глюкоза-6-фосфата, также может покидать гликолиз и становиться участником биосинтеза гексозаминов в качестве субстрата. Первым шагом в биосинтезе гексозаминов является катализируемая глутамин-фруктоза-6-фосфат-аминотрансферазой 1 (glutamine fructose-6-phosphate aminotransferase 1, GFPT1) реакция образования глюкозамин-6-фосфата из фруктоза-6-фосфата и глутамина. Благодаря образованию N-ацетилглюкозамина (GlcNAc) — субстрата для N- и О-гликозилирования — гексозаминовый путь обеспечивает клетку субстратами для реакций гликозилирования, а также для биосинтеза гепарансульфата и гиалуроновой кислоты. Кроме этого, он потенцирует работу рецептор-опосредованных сигнальных путей и регулирует стабильность таких белков, как c-myc.
Следующий интермедиат гликолиза, используемый в синтезе макромолекул, — это дигидроксиацетонфосфат (ДГАФ). ДГАФ под действием глицерол-3-фосфатдегидрогеназы 1 (glycerol-3-phosphate dehydrogenase 1, GPD1) превращается в глицерол-3-фосфат, который участвует в биосинтезе различных фосфолипидов, что представляют собой важнейший структурный компонент клеточной мембраны.
Вероятно, наиболее интенсивно изучаемым рост-стимулирующим механизмом, переводящим метаболиты гликолиза в другие пути, является участие 3-фосфоглицерата в качестве предшественника для биосинтеза серина и глицина, а также в образовании метильных донорных групп и НАДФН. Согласно работам по изучению метаболических путей в организме, опухолевые клетки могут использовать до 50% углерода, полученного от глюкозы, в биосинтезе и дальнейшем катаболизме серина. Фермент 3-фосфоглицератдегидрогеназа (3-phosphoglycerate dehydrogenase, PHGDH), лимитирующий скорость биосинтеза серина, зачастую локально амплифицирован при раке молочной железы и меланоме и необходим для роста клеток с амплифицированной PHGDH in vitro и in vivo.
Серин играет уникальную роль в метаболизме, будучи главным субстратом для так называемого одноуглеродного, или фолатного цикла. На его последних этапах углерод серина в гамма-положении может переноситься на особую молекулу-носитель – тетрагидрофолат (ТГФ). В ходе реакции, катализируемой сериновой гидроксиметилтрансферазой 2 (СГМТ-2) в митохондриях и СГМТ-1 – в цитозоле, образуются 5,10-метилен-ТГФ и глицин. Далее 5,10-метилен-ТГФ претерпевает серию окислительно-восстановительных преобразований, в результате чего образуется линейка ТГФ, несущих одноуглеродные фрагменты. Такие молекулы ТГФ используются в качестве субстратов для биосинтеза пуринов, тимидина, а также для образования S-аденозилметионина – важнейшего субстрата для клеточных реакций метилирования. Кроме этого, было показано, что поэтапное окисление несущих одноуглеродные группы ТГФ позволяет получить до 50% всех клеточных молекул НАДФН. Также выявлено, что опосредованное гипоксией повышение экспрессии СГМТ-2 защищает клетки от оксидативного стресса, ассоциированного с состоянием гипоксии. Таким образом, метаболиты одноуглеродного пути участвуют во множестве биосинтетических и регуляторных процессов в клетке. Наконец, метилентетрагидрофолатдегидрогеназа-2 (МТГФД-2), компонент митохондриальной ветки одноуглеродного пути, признаётся одним из трёх наиболее часто сверхэкспрессированных метаболических ферментов, встречающихся в злокачественных опухолях. Это приводит к мысли, что повреждения в фолатном пути могут специально отбираться и сохраняться в ходе опухолеобразования.
В заключение, интермедиаты гликолитического пути могут покидать гликолиз и принимать участие в различных биосинтетических процессах; в связи с этим, скорость-лимитирующие ферменты путей-ответвлений от гликолиза зачастую представлены в опухолях в сверхэкспрессированном виде. Для поддержания баланса между конечным выходом от гликолиза и его ролью в образовании пирувата, поддерживающего активность ЦТК, активно пролиферирующие клетки разработали новый особый механизм, который регулировал бы последний этап гликолиза. Этот этап контролируется пируваткиназой (ПК) – ферментом, конвертирующим фосфоенолпируват в собственно финальный продукт гликолиза, пируват. За исключением печени и почек, где экспрессируются тканеспецифичные изоформы ПК (ПК типов L и R), большинство тканей экспрессируют М-форму ПК (мышечную). ПКМ существует в двух сплайсируемых вариантах. Несмотря на то, что ПКМ-1 является наиболее эффективным в образовании пирувата, подавляющая часть быстро пролиферирующих клеток и практически все опухолевые клетки экспрессируют преимущественно ПКМ-2.
По сравнению с ПКМ-1, активность ПКМ-2 находится под строгой регуляцией. Так, ПКМ-2 ингибируется тирозиновым фосфорилированием и активируется – сериновым. Таким образом, зависимая от факторов роста сигнальная трансдукция ингибирует ПКМ-2, что ведёт к накоплению гликолитических промежуточных метаболитов до тех пор, пока не удовлетворятся нужды растущей клетки в пуле свободного серина. В поддержку этой парадигмы можно отметить, что введение в игру изоформы ПКМ-1 нарушает пролиферацию опухолевых клеток и опухолеобразование in vivo, в частности благодаря негативному влиянию на серин-зависимые биосинтетические процессы. Помимо серина, ПКМ-2 может также аллостерически активироваться избытком других накопленных промежуточных продуктов метаболизма глюкозы, включая сукциниламиноимидазолкарбоксамид (САИКАР) – интермедиат биосинтеза рибоза-5-фосфата.
Появляющиеся научные свидетельства говорят о том, что гликолиз используется делящимися клетками в качестве гибкого и изменчивого механизма, поставляющего метаболические интермедиаты для многочисленных биосинтетических процессов. Любые избытки продуктов гликолиза, не использованные для биосинтеза, преимущественно конвертируются в лактат для сохранения необходимого пула НАД+ в целях поддержания гликолиза и избегания заполнения митохондрий молекулами НАДН, которые подавляли бы ЦТК. Избыток НАДН, образованный в ходе гликолиза, может перемещаться в митохондрии интенсивно пролиферирующих клеток посредством химических переносчиков или использоваться для превращения ДГАФ, интермедиата гликолиза, в глицерол-3-фосфат, который поставляет электроны напрямую в электрон-транспортную цепь. В этой реакции участвует глицерол-3-фосфатдегидрогеназа (мГФДГ), заякоренная на наружней мембране митохондрий. Благодаря задействованию ФАД в качестве акцептора электронов, мГФДГ-катализируемая реакция снижает митохондриальный электрохимический потенциал НАДН, образованного в ходе гликолиза. Известно, что активность ГФДГ повышена в клетках рака простаты, инсулиномах и карциноидных опухолях.
Митохондрии защищены от избыточного гликолиза также тем, что любой пируват, вступающий в ЦТК, переводится в цитрат, который может быть выброшен «за борт» трикарбоксилатным переносчиком в цитозоль и расщеплён до ацетил-КоА и оксалоацетата. Оксалоацетат превращается в малат и обратно перемещается в митохондрии в целях анаплероза, а ацетил-КоА служит предшественником для биосинтеза липидов и ацетилирования белков. Электроны, поступающие в митохондриальную электрон-транспортную цепь благодаря активности глицеролфосфатдегидрогеназы и Комплекса I, полученные в ходе превращения малата в оксалоацетат, достаточны для обеспечения митохондриальной целостности и синтеза АТФ, несмотря на снижение катаболической активности ЦТК.
Было продемонстрировано, что онкогены способны приводить к описанным выше адаптациям для быстро делящихся клеток. К примеру, с-myc повышает экспрессию: ПДК-1; лактатдегидрогеназ типа А (ЛДГ-А), катализирующих восстановительную реакцию превращения пирувата в лактат; а также монокарбоксилатного транспортного белка (МКТ-1), способствующего выбросу лактата во внеклеточное пространство. Помимо с-myc, транскрипционная активность МКТ-1 и ПДК-1 повышается также благодаря бета-катенин/ТКФ-сигнальному пути. В конце концов, стабилизация молекулы HIF1α в состоянии гипоксии или различными онкогенами также вызывает повышение транскрипционной активности ЛДГ-А и ПДК-1.
Даже при наличии этих адаптаций, быстро пролиферирующие клетки зачастую аккумулируют электрон-транспортный поток частиц в объёмах, превышающих мощность АТФ-синтазы, что приводит к формированию избытка активных форм кислорода (АФК). Более того, последствия повреждения от перепроизводства АФК, вызванные перегруженной электрон-транспортной цепью, могут стать причиной феномена онкоген-индуцированного клеточного старения (ОИС). Вызванное внесением мутантных аллелей BRAF или RAS, ОИС ассоциировано с глубоким окислительным повреждением и необратимым прекращением роста. Интересно, что ослабление активности пируватдегидрогеназы (ПДГ), фермента-«контроллёра», конвертирующего пируват в ацетил-КоА, – при условии эктопической экспрессии его негативного регулятора пируватдегидрогеназной киназы 1 (ПДК-1) или иРНК-опосредованной деплеции ПДГ-фосфатазы ПДФ-2 – доказанно «обходит» индуцированное мутантным BRAFV600E ОИС и усиливает опухолеобразование.
В то время как перепроизводство АФК оказывает губительное влияние на клеточный рост и выживаемость, умеренные количества АФК, образованные в результате тканевого дыхания или целенаправленно с участием НАДФ-оксидаз, вносят важный вклад в метаболический сигналинг, ведущий к опухолевому перерождению. В этой связи, АФК действуют как ингибиторы протеинфосфатаз, таких как PTEN и PTP1B, а также активаторов киназного семейства Src и МАРК. Более того, увеличение уровня продукции АФК способствует активации транскрипционных факторов HIF1α и NRF2, что усиливает транскрипционные программы, ведущие в дальнейшем к опухолеобразованию.
По сравнению с пролиферирующей основой опухоли, те её субпопуляции клеток, что находятся в спящем состоянии, согласно научным данным, значительно менее гликолитически активны и испытывают гораздо большую зависимость от окислительного фосфорилирования, а также от повышенной экспрессии митохондриальных компонентов тканевого дыхания. Среди таких популяций находятся также и стволово-подобные субклоны, которые возникают после онкогенной абляции in vivo, а кроме них — циркулирующие опухолевые клетки. Подобный дихотомический принцип в использовании углерода для преимущественно биосинтетических, или же преимущественно биоэнергетических целей объясняет, почему опухоли сохраняют потенциал для окислительного фосфорилирования, в отличие от оригинальной гипотезы Отто Варбурга.
Наряду с интермедиатами гликолиза, сигнальный путь факторов роста повышает темпы расходования отдельных промежуточных метаболитов ЦТК в процессе образования биосинтетических предшественников. В частности, активация PI3K/Akt-пути позволяет клетке нарастить цитозольный пул молекул ацетил-КоА — субстрата для биосинтеза жирных кислот de novo. Это происходит благодаря Akt: эта киназа активирует АТФ-зависимую цитратлиазу (АЦЛ), катализирующую цитозольное расщепление цитрата, поступившего из ЦТК, до ацетил-КоА и оксалоацетата.
В то время как темпы биосинтеза жирных кислот de novo в нормальных клетках взрослого организма довольно низки — за исключением таких липогенных тканей, как печень, жировая ткань, эпителий молочных желёз в период лактации, — опухолеобразование ассоциировано со значительным увеличением образования липидов. Повышенная способность синтеза липидов de novo не только способствует формированию новых участков липидного бислоя, но также даёт клетке возможность преобразовывать свою мембрану с увеличением содержания в ней устойчивых к окислительному повреждению насыщенных жирных кислот, что помогает адаптироваться к оксидативному стрессу. Биосинтез цепей жирных кислот начинается с карбоксилирования цитозольных ацетил-КоА ферментом ацетил-КоА-карбоксилазой (АКК), что ведёт к образованию малонил-КоА, молекулы которых далее объединяются в цепи длинных жирных кислот при участии синтазы жирных кислот (СЖК). Что интересно, все три главные компоненты, вовлечённые в биосинтез цепей жирных кислот — АЦЛ, АКК и СЖК — зачастую сверхэкспрессированы в трансформированных клетках; более того, их ингибирование ослабляет рост опухолевых клеток in vitro и in vivo. Помимо образования жирных кислот, значительную роль в опухолеобразовании играет также и биосинтез холестерола de novo из малонила-КоА. Так, например, препятствие биосинтезу холестерола путём ингибирования соответствующего скорость-лимитирующего фермента, гидроксиметилглутарил-КоА-редуктазы (ГМГ-КоА-редуктазаы), сохраняет нормальную ацинарную морфологию клеток рака молочной железы. Это означает, что изменения в структуре и текучести мембраны может влиять на архитектуру ткани и свободный, «безъякорный» клеточный рост.
В дополнение к поставке цитрата, ЦТК также обеспечивает клетки метаболическими предшественниками для биосинтеза заменимых аминокислот, включая аспартат и аспарагин. Недавно стало известно, что биосинтез аспартата достаточно сильно зависит от способности клетки к окислительному фосфорилированию. Для того чтобы синтез этих аминокислот постоянно поддерживался, должен соблюдаться тонкий баланс между их образованием и введением в цикл анаплеротических метаболитов. Главным анаплеротическим субстратом в растущих клетках является глутамин. Глутамин может захватываться митохондриями при участии глутаминазы; образованный в ходе реакции глутамат под действием глутаматдегидрогеназы или трансаминаз превращается в α-кетоглутарат. Большинство быстро пролиферирующих клеток зависят от постоянных поставок глутамина для сохранения интегральной целостности промежуточных метаболитов ЦТК. В случае с c-myc-опосредованно трансформированными клетками, недостаток глутамина ведёт к коллапсу ЦТК и вызывает клеточную гибель, что может предотвратить добавление оксалоацетата или проникающей через мембрану форму α-кетоглутаровой кислоты. Окисление полученного от глутамина α-кетоглутарата до оксалоацетата не только помогает сохранить способность клеток к синтезу цитрата. Кроме этого, образующийся оксалоацетат может быть конвертирован в малат, который, в свою очередь, может далее окисляться малик-энзимом (МЭ-1) до пирувата с образованием НАДФН, что протекает в некоторой степени независимо от глюкозы. В конечном итоге, α-кетоглутарат может обеспечивать пул цитрата для липогенеза de novo в условиях дефицита образованного от катаболизма глюкозы ацетила-КоА, что может быть вызвано гипоксией или действием некоторых онкогенов. В процессе, именуемом восстановительным карбоксилированием, часть реакций ЦТК начинает идти в обратном направлении, что ведёт к образованию цитрата из α-кетоглутарата. Цитрат далее может использоваться для образования цитозольного пула молекул ацетил-КоА.
Полученная от глутамина α-кетоглутаровая кислота выступает не только лишь в качестве анаплеротического субстрата, который может использоваться быстро пролиферирующими клетками. Кстати, благодаря отслеживанию судьбы углеродных элементов — метаболитов гликолиза — у пациентов и у мышиных моделей с глиобластомой и немелкоклеточным раком лёгкого (НМРЛ) удалось заметить, что как минимум в этих двух случаях в качестве главного ресурса для ЦТК выступала глюкоза, а не глутамин. Анаплеротический вход производных от глюкозы углеродных соединений в ЦТК имеет место благодаря карбоксилированию пирувата с образованием оксалоацетата (интермедиата ЦТК), что катализируется пируваткарбоксилазой (ПК). Более того, было продемонстрировано, что выживаемость устойчивых к дефициту глутамина потомков клеточной линии глиобластомы в культуре зависит от пируваткарбоксилазы. Помимо этого, ингибиторы ПК в клеточных линиях НМРЛ угнетают биосинтез жирных кислот и значительно снижают пролиферацию даже в условиях избытка глутамина. Эти исследования иллюстрируют, что важную роль при выборе анаплеротического субстрата играет как конкретный вид окружающей клеточной линии, так и потенциальные вторичные эффекты, связанные с адаптацией клеток к условиям культуры ткани.
В заключение, внеклеточный ацетат, как недавно было выяснено, используется некоторыми опухолями в качестве ресурса для биосинтеза ацетил-КоА. Например, различные типы опухолей, локализованные в мозге, включая первичные глиобластомы, ассимилируют экзогенный ацетат и встраивают полученные от него углеродные продукты в процесс синтеза жирных кислот de novo. Поглощённый ацетат конвертируется в ацетил-КоА в ходе реакции, катализируемой ацетил-КоА-синтетазой-2 (АКС-2) — мишенью для амплификации в раке молочной железы. Таким образом, ассоциированный с гипоксией дефицит ацетил-КоА может усиливаться не только посредством карбоксилирования α-кетоглутаровой кислоты, но и за счёт повышения потребления свободного ацетата из плазмы и межклеточной жидкости. Более того, повышение экспрессии АКС-2 потенциально может позволять клеткам перерабатывать ацетат, полученный в ходе деацетилирования гистонов и других клеточных белков.
Повышенная потребность в азоте
Помимо повышения объёма поглощения углеродных соединений для биосинтетических процессов, ростовой сигнальный путь одновременно усиливает потребность клетки в восстановленных соединениях азота. Так, быстро пролиферирующей клетке приходится синтезировать какое-то количество азотосодержащих молекул de novo, включая нуклеотиды, заменимые аминокислоты и полиамины. Как отмечалось выше, транскрипция c-myc и E2F ведёт к усилению клеточного потребления глутамина. Глутамин — заменимая аминокислота, содержащая два атома азота в восстановленной форме — служит, главным образом, в качестве «разменной монеты», благодаря которой азот в восстановленной форме перемещается между клетками в организмах многоклеточных животных. Амидная группа глутамина является незаменимым донором азота для биосинтеза пуриновых и пиримидиновых оснований (Рисунок 5). Так, в образовании урацила и тимина участвует по одной молекуле глутамина, а цитозин и аденин требуют по две; построение гуанинового основания стоит клетке трёх молекул глутамина. Помимо этого, для пиримидинового и пуринового колец необходим аспартат, получаемый посредством трансаминирования метаболита ЦТК оксалоацетата и глутаминовой кислоты (оба — катаболиты глутамина). Таким образом, глутамин является ключевым структурным строительным элементом в биосинтезе нуклеотидов. Кроме этого, согласно исследованиям, концентрация глутамина определяет скорость прохождения клеточного цикла, а дефицит глутамина ведёт в определённых случаях к задержке клеточного цикла в S-фазе.
Онкоген c-myc руководит биосинтезом нуклеотидов посредством повышения экспрессии некоторых соответствующих ферментов биосинтеза. Среди c-myc-регулируемых мишеней различают фосфорибозилфосфатсинтетазу-2 (ФРФС-2), катализирующую первый этап биосинтеза пуринов, а также кабамоилфосфатсинтетазу II (КФС-2), запускающую каскад образования пиримидинового кольца. Другие мишени c-myc, вовлечённые в биосинтез нуклеотидов, — это тимидилатсинтаза (ТС) и инозинмонофосфат-дегидрогеназа-1 (ИМФДГ-1) и -2 (ИМФДГ-2). В связи с этим, c-myc не только способствует захвату глутамина, но также усиливает его утилизацию в ходе биогенеза пуриновых и пиримидиновых оснований. Вдобавок к регуляторному вкладу со стороны c-myc, на экспрессию генов биосинтеза нуклеотидов влияют также мутантные аллели р53, среди мишеней которых отмечены ИМФДГ-1, ИМФДГ-2, ГМФ-синтетаза, а также ферменты нуклеозидной реутилизации деоксицитидинкиназа (ДЦК) и тимидинкиназа (ТК-1). Более того, активность фермента КФС-2 регулируется посредством фосфорилирования МАРК и благодаря mTORC1-зависимой S6-киназе. Похожим образом, mTORC1-опосредованная активация КФС-2 способствует клеточному регулированию биогенеза пиримидинов в ответ на конкретную внутриклеточную концентрацию глутамина, при которой происходит активация mTORC1.
Помимо участия в биосинтезе нуклеотидов, глутамин может напрямую дезаминироваться в глутамат при участии глутаминазы, сверхэкспрессия которой зачастую наблюдается в опухолевых клетках под влиянием c-myc. Образованный таким путём глутамат служит в качестве донора азота для образования некоторых заменимых аминокислот в результате процесса трансаминирования. Напротив, для биосинтеза аспарагина из аспартата, катализируемого аспарагинсинтетазой (АСС), требуется амидный азот глутамина. Несмотря на структурное сходство с глутамином, аспарагин — это единственная аминокислота, которая не катаболизируется клетками млекопитающих. Вместе с тем, аспарагин играет важнейшую регуляторную роль в условиях дефицита глутамина. То, как именно аспарагин способствует клеточному выживанию и адаптации к нехватке глутамина, ещё предстоит выяснить. АСС зачастую сверхэкспрессирована в опухолях и это ассоциировано с неблагоприятным прогнозом. С другой стороны, клетки острого лимфобластного лейкоза не экспрессируют АСС, что обуславливает их ауксотрофию по аспарагину. Снижение концентрации аспарагина в плазме под действием рекомбинантной формы бактериальной L-аспарагиназы признано эффективным противоопухолевым лечением для этого типа неоплазии.
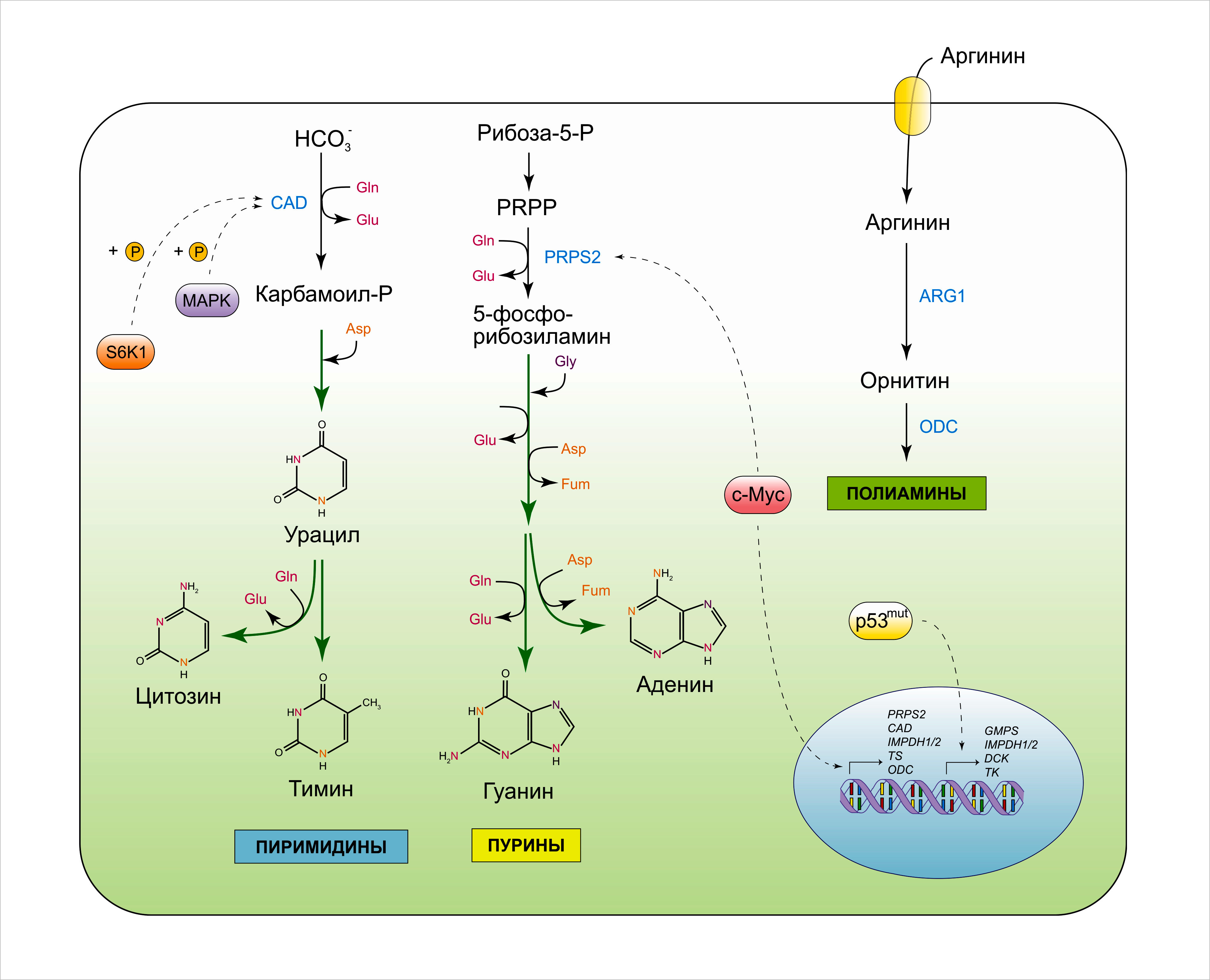
PRPP, фосфорибозилпирофосфат; PRPS2, фосфорибозилпирофосфат-синтетаза 2; CAD, карбамоилфосфат-синтетаза 2; Gln, глутамин, Glu, глутамат; Gly, глицин; Asp, аспартат; Fum, фумарат; IMPDH1/2, инозин-5-монофосфат-дегидрогеназа; TS, тимидилатсинтаза; GMPS, гуанозинмонофосфат-синтетаза; DCK, деоксицитидинкиназа; TK, тимидинкиназа, ARG1, аргиназа 1; ODC, орнитиндекарбоксилаза; CAT1/2, переносчик катионных аминокислот.
Несмотря на наличие в клетках млекопитающих полноценного пути глутаминового биосинтеза, большинству быстро пролиферирующих клеток в культуре необходимо экзогенное поступление глутамина. Интересно, что некоторые клеточные типы, такие как эмбриональные стволовые клетки и клетки люминального рака молочной железы, способны к пролиферации в отсутствие глутамина в культурной среде, что указывает на возможное образование глутамина de novo при определённых обстоятельствах. Кроме этого, было обнаружено, что в человеческих образцах глиобластомы, имплантированных напрямую в мозг мышей с ослабленным иммунитетом, аккумулируется объём глутамина, сходный с таковой в нормальной ткани мозга; более того, схожая аккумуляция, произведённая от глюкозы глутамина в опухолевой ткани, наблюдалась в мышиной модели c-Met-опосредованного рака печени. Глутаминсинтаза (ГС) сверхэкспрессирована в некоторых опухолях, но её роль и механизм активации ещё предстоит изучить.
Аргинин, хоть и является заменимой аминокислотой, становится условно-незаменимой в некоторых ситуациях опухолегенного характера. Будучи переносчиком четырёх атомов азота, аргинин служит предшественником для разнообразных азотсодержащих элементов, включая полиамины, креатин, агматин и пирролин-5-карбоксилат — предшественник биосинтеза пролина. Биосинтез аргинина de novo является неотъемлемой частью цикла мочевинообразования, куда он вступает при участии аргининосукцинат-лиазы (АСЛ), катализирующей реакцию расщепления аргининосукцината. Аргининосукцинат, в свою очередь, образуется благодаря ферменту аргининосукцинатсинтазе (АрСС-1) из цитруллина и аспартата. Любопытен тот факт, что как АрСС-1, так и АСЛ зачастую находятся под эпигенетическим сайленсингом в клетках меланомы, почечно-клеточного рака и гепатоцеллюлярной карциномы. Сниженная экспрессия АрСС-1 и АСЛ ассоциирована с неблагоприятным прогнозом и устойчивостью к химиотерапии. В соответствии с этим, данные об ауксотрофии опухолей по аргинину уже приняты к сведению для разработки противоопухолевых тактик лечения.
Почему опухоли отказываются от образования аргинина de novo и, вместо этого, начинают полагаться лишь на его экзогенное поступление? Одним из возможных ответов является то, что инактивация АСЛ и АрСС-1 позволяет опухолевым клеткам накапливать орнитин, используемый далее для синтеза полиаминов — класса азотсодержащих поликатионных алифатических углеродных молекул. Концентрация полиаминов увеличивается в быстро пролиферирующих клетках; более того, было продемонстрировано, что полиамины ингибируют апоптоз и усиливают опухолевый рост и инвазию. Другая возможная причина для ауксотрофии по аргинину в опухолях может быть связана с тем фактом, что подавление АрСС-1-опосредованного образования аргининосукцината ведёт к накоплению его субстрата — аспартата, который может вступать в путь биосинтеза нуклеотидов.
Вдобавок ко влиянию на метаболизм глутамина, c-myc-контролируемое ремоделирование метаболизма заменимых аминокислот включает в себя метаболизм пролина. Пролин может синтезироваться как из глутамата, так и из полученного от аргинина орнитина через общий промежуточный метаболит — пирролин-5-карбоксилат. Экспрессия ключевого фермента биосинтеза пролина, пирролин-5-карбоксилат-редуктазы (П5КР-1), повышается благодаря c-myc. Что интересно, мета-анализ экспрессии метаболических ферментов идентифицировал П5КР-1 как один из наиболее часто сверхэкспрессированных генов в опухолях. Соответственно, пролин-оксидаза (ПО), осуществляющая деградацию пролина, ингибируется c-myc посредством miR-23b, а также является мишенью р53. Было продемонстрировано, что экспрессия ПО подавляет рост опухолевых клеток, вызывая задержку клеточного цикла на фазе G2. Вклад нарушенного метаболизма пролина в опухолеобразование ещё предстоит изучить подробнее. Одно из объяснений предполагает, что накопление пролина может способствовать синтезу коллагена и формированию новых элементов внеклеточного матрикса, что, таким образом, содействует опухолевой инвазии. В целом, хоть эта сфера опухолевого метаболизма всё ещё продолжает активно изучаться, уже можно утверждать, что метаболизм азота в ходе опухолеобразования претерпевает, как и в случае с углеродом, сложные перестройки.
Нарушения в генной регуляции, опосредованной метаболитами
Ошибочно активированные сигналы роста и выживания, инициирующие опухолеобразование, способствуют перестройке метаболизма опухолевых клеток в сторону увеличения захвата питательных веществ и наращивания темпов биосинтеза. Однако метаболические сети не только лишь являются пассивными реципиентами ростовых сигналов, но и сами напрямую передают информацию о состоянии клеточного метаболизма различным регуляторным ферментам, среди которых и те, что опосредуют перестановку и снятие эпигенетических маркёров с хроматина (Рисунок 6).
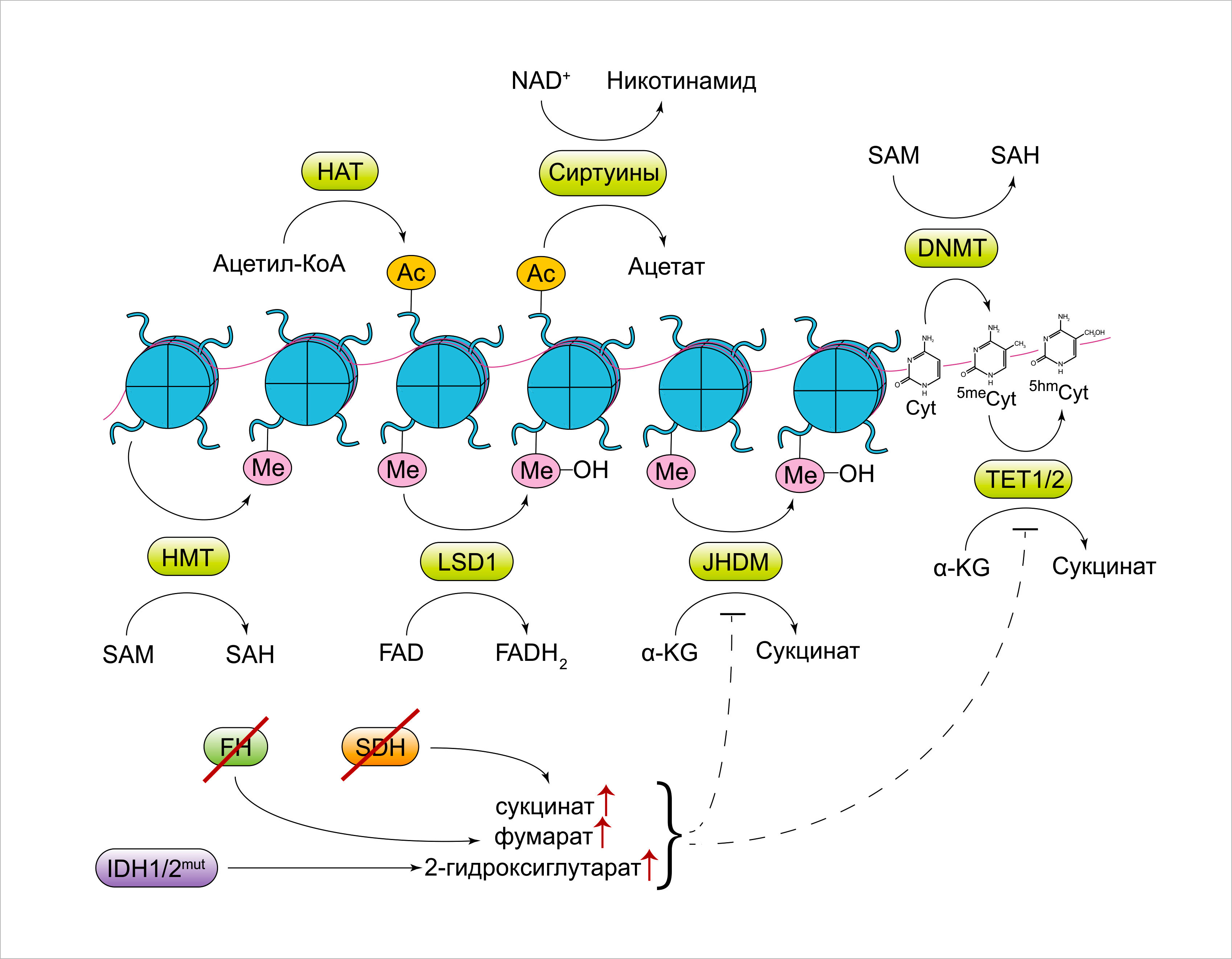
HAT, гистоновые ацетилтрансферазы; Ac, ацетильный маркёр; SAM, S-аденозилметионин; SAH, S-аденозилгомоцистеин; DNMT, ДНК-метилтрансферазы; HMT, гистоновые метилтрансферазы; Me, метильный маркёр; LSD1, лизин-специфическая гистоновая деметилаза 1; JHDM, гистоновые деметилазы, содержащие домен Jumonji; Cyt, цитозин; 5meCyt, 5-метилцитозин; 5hmCyt, 5-гидроксиметилцитозин; TET1/2, ten-eleven транслокационные метилцитозиновые диоксигеназы 1/2; a-KG, a-кетоглутарат; SDH, сукцинатдегидрогеназа; FH, фумаратгидратаза; IDH1/2, изоцитратдегидрогеназа 1/2.
Ключевым метаболитом, синтезирующемся при более избыточном метаболизировании глюкозы клеткой, чем это необходимо для биоэнергетических нужд, является цитозольная фракция ацетил-КоА. Цитозольный ацетил-КоА считается незаменимым субстратом для ферментов, осуществляющих ацетилирование гистонов и других белков. «Навешивание» ацетильных остатков на гистоны ассоциировано с повышением доступности геномной ДНК для механизма сборки транскрипционных комплексов; эта связь в норме недолговечна. Ацетилирование гистонов чрезвычайно чувствительно к повреждениям в клеточном трофическом и сигнальном статусах. Примечательно, что прекращение поставки и внесение глюкозы обратно, как и активация онкогенного сигнального пути посредством включения онкогенного KRAS мутантной или перманентно активной формой Akt, повышает общую долю ацетилированных гистонов, что, в свою очередь, ведёт к усилению генной экспрессии. Иммуногистохимический анализ образцов глиобластомы и опухоли простаты показал, что уровень активации Akt коррелирует с общей долей ацетилирования. Активированные Akt увеличивают экстрамитохондриальный пул молекул ацетил-КоА путём активирования АЦЛ, конвертирующую цитозольный цитрат в ацетил-КоА и оксалоацетат. Более того, было выявлено, что АЦЛ подавляет как глюкозо-, так и онкоген-опосредованное ацетилирование гистонов.
Помимо ацетил-КоА, гистонацетилтрансфераза р300 использует в качестве субстрата также и молекулы кротонил-КоА. Примечательно, что перестановка кротонильных маркёров на определённые аминокислотные остатки в гистоновых хвостах активирует экспрессию генов даже в большей степени, чем ацетильные маркёры. Кротонил-КоА может синтезироваться в ходе катаболизма лизина и триптофана, а также в ходе распада короткоцепочечной жирной кислоты бутирата. Кстати, многие новые виды модификации гистонов, такие как формилирование, пропионилирование, бутирилирование, малонилирование и сукцинилирование были идентифицированы благодаря тандемной масс-спектрометрии. Дальнейшие исследования этих маркёров поможет расширить данные о количестве метаболических факторов, регулирующих глобальные паттерны экспрессии генов.
В многочисленных реакциях метилирования в клетке, включая перестановку метильных остатков на гистоновых хвостах, метилирование цитозина на ДНК и аденина — на мРНК, используется S-аденозилметионин (SAM) в качестве донора метильных группировок. SAM является продуктом одноуглеродного метаболического пути и участвует в катаболизме серина, как описано выше. Множество новых работ демонстрируют, что реакции метилирования гистонов и ДНК чувствительны к концентрации SAM.
Снятие ацетильных и метильных меток также находится под регуляцией клеточного состояние метаболизма. К примеру, сиртуины, принадлежащие к классу деацетилаз и катализирующие отщепление ацетильной группы от гистоновых и других белков, используют в качестве кофактора NAD+, в то время как FAD участвует как кофактор лизин-специфической деацетилазы ЛСД1. Будучи чувствительными к доступности NAD+ и FAD, эти три фермента управляют глобальными посттрансляционными и эпигенетическими изменениями, стимулирующими сохранение энергии.
Различные посттрансляционные модификации в клетке осуществляются членами большого класса зависимых от присутствия α-кетоглутарата диоксигеназ. Среди α-кетоглутарат-зависимых диоксигеназ различают ДНК-деметилазы из семейства ТЕТ, гистоновые деметилазы семейства Jumonji C, мРНК-деметилазы FTO и ALKBH5, а также семейство пролил-гидроксилазных (ПГ) ферментов, которые, среди всего прочего, регулируют концентрацию HIF1a в ответ на колебания кислорода и оксидативный стресс. Механизм действия α-кетоглутарат-зависимых диоксигеназ включает одновременное оксиление ко-субстрата α-кетоглутарата до янтарной кислоты. Соответственно, внутриклеточная концентрация α-кетоглутарата может напрямую влиять на активность этих ферментов. Кроме этого, кетоглутарат-зависимые диоксигеназы подвержены ингибированию продуктами своей же реакции — сукцинатом и фумаратом, образующимся в ходе ЦТК из сукцината. К примеру, потеря обоих аллелей сукцинатдегидрогеназы (СДГ), что можно наблюдать при семейных параганглиомах и феохромоцитомах, а также в случае ряда спорадических гастроинтестинальных стромальных опухолей, ведёт к накоплению сукцината. Схожим образом, потеря фумарат-метаболизирующего фермента фумарат-гидратазы (ФГ) и накопление фумарата было продемонстрировано при семейном синдроме НЛПКР (наследственного лейомиоматоза и почечно-клеточного рака), а также в ряде случаев параганглиом и феохромоцитом. Опухоли с потерей СДГ и ФГ имеют схожие фенотипические особенности, согласующиеся с подавлением диоксигеназ, среди которых можно отметить характерную повышенную активность метилирования ДНК, а также возрастание концентрации молекул HIF1a. Интересно, что последнее, как минимум в контексте потери СДГ, может полностью измениться добавлением проникающей через мембрану формы α-кетоглутарата.
Другим выдающимся классом опухоль-ассоциированных генетических нарушений, повышающих активность α-кетоглутарат-зависимых диоксигеназ, являются мутации с приобретением функции (gain-of-function mutations) изоцитратдегидрогеназы-1 (ИДГ-1) и -2 (ИДГ-2). Мутации в ИДГ-1 и -2 идентифицированы в образцах глиомы низкой степени злокачественности, где они отображают местонахождение вероятного первичного очага, а также при хондросаркоме, холангиокарциноме и остром миелолейкозе (ОМЛ). Мутантные аллели ИДГ-1 и -2 обладают необычной неоморфной ферментативной активностью. По сравнению с изоцитрат-дегидрогеназами дикого типа, конвертирующими метаболит ЦТК изоцитрат в α-кетоглутаровую кислоту, мутантная форма ИДГ предпочитает использовать α-кетоглутарат в качестве субстрата, катализируя его превращение в D-энантиомер 2-гидроксиглутарата (2-ГГ). Благодаря структурной схожести с α-кетоглутаратом, 2-ГГ участвует как конкурентный ингибитор α-кетоглутарат-зависимых диоксигеназ. Примечательно, что ИДГ-опосредованные глиомы, лейкозы и хондросаркомы имеют выдающийся СрG-участок гиперметилирования, что напоминает тот же фенотип в случаях СДГ- ФГ-дефицитных опухолей. Мутации ИДГ при ОМЛ несовместимы с инактивирующими мутациями ТЕТ2-метилцитозин-гидроксилаз, в дальнейшем вовлекающие ИДГ в качестве надёжного драйвера эпигенетических перестроек. Более того, включение мутантного аллеля ИДГ-1 в нормальных гематопоэтических клетках и в хондроцитах in vivo вызывает аномальную экспансию целевых линий клеточной дифференцировки. Среди различных клеточных параметров, эктопическая экспрессия мутантного аллеля ИДГ или лечение экзгоненным D-2-ГГ достаточны для активации гиперметилирования ДНК и гистонов и для блокирования клеточной дифференцировки. С другой стороны, опосредованное малыми молекулами ингибирование мутантной ИДГ-1 в глиомах и ИДГ-2 при лейкозе существенно усиливает дифференцировку. Избыток 2-ГГ в клетке можно зафиксировать благодаря ЯМР-спектроскопии, что уже применяется при изучении мутантных по ИДГ глиом. Более того, малые молекулы-ингибиторы ИДГ-1 и ИДГ-2, согласно научным данным, вызывают ремиссии в фазе I клинических испытаний при лечении ОМЛ у пациентов с соответствующими мутациями.
Примечательно, что некоторые типы опухолей демонстрируют повышенные уровни 2-ГГ даже в отсутствие мутаций ИДГ. К примеру, повышенные концентрации 2-ГГ обнаружены в случаях тройного негативного рака молочной железы. Помимо этого, специфическое возрастание уровня L-энантиомера 2-ГГ, проявляющего сходный, но не такой же ингибиторный эффект на α-кетоглутарат-зависимые диоксигеназы, было обнаружено при светлоклеточном раке почки, где оно коррелирует с повышенной концентрацией метилцитозина. Вдобавок, было показано, что уровень L-2-ГГ в клетке увеличивается в ответ на гипоксию, проявляющуюся в результате использования ферментами ЛДГ-А и малат-дегидрогеназой (МДГ) нерегулярных, неспецифических субстратов. В двух недавних исследованиях молекулы L-2-ГГ описываются в качестве метаболических медиаторов клеточного антистрессового ответа на гипоксию.
Метаболические взаимодействия с микроокружением
Информация о метаболическом состоянии клетки не только влияет на её поведение в будущем, но также может воздействовать на судьбу других клеток по соседству. Так, ряд генетически стабильных клеточных типов, среди которых — ассоциированные с опухолью фибробласты, эндотелиальные клетки, а также компоненты врождённого и приобретённого иммунитета, известны своей способностью претерпевать характерные фенотипические изменения в ответ на «расположившуюся» по соседству прогрессирующую опухоль. В сфере интенсивного изучения на данный момент находится вопрос о том, как опухолевые клетки преобразуют своё микроокружение для способствования опухолевому росту и диссеминации; однако уже сейчас известно, что подобные тактики перепрограммирования своей среды охватывают сразу несколько стратегий, среди которых — секреция факторов роста, перестройки внеклеточного матрикса и нарушения межклеточных взаимодействий.
Помимо этого, быстро пролиферирующие опухолевые клетки повреждают также и метаболическую структуру внеклеточной среды вокруг себя (Рисунок 7). Повышенные темпы утилизации внеклеточной глюкозы и глутамина опухолевыми клетками ведёт к накоплению внеклеточного лактата, что, как оказалось, влияет на многочисленные клеточные типы внутри опухолевого микроокружения. Возрастание уровня лактата ведёт к образованию малочувствительного к иммунному ответу микроокружению благодаря ослаблению активации дендритных и Т-клеток, а также миграции моноцитов. Помимо этого, лактат стимулирует поляризацию резидентных макрофагов в так называемое «состояние М2», что играет важную роль в иммуносупрессии и заживлении ран. Кроме того, накопление лактата усиливает темпы ангиогенеза. Таким образом, лактат поддерживает в стабильном состоянии HIF1a и активирует сигнальные пути NF-kB и PI-3-киназы в эндотелиальных клетках, а также способствует секреции проангиогенного фактора VEGF из ассоциированных с опухолью стромальных клеток. Возрастание концентрации лактата также стимулирует образование фибробластами гиалуроновой кислоты, что может способствовать опухолевой инвазии.
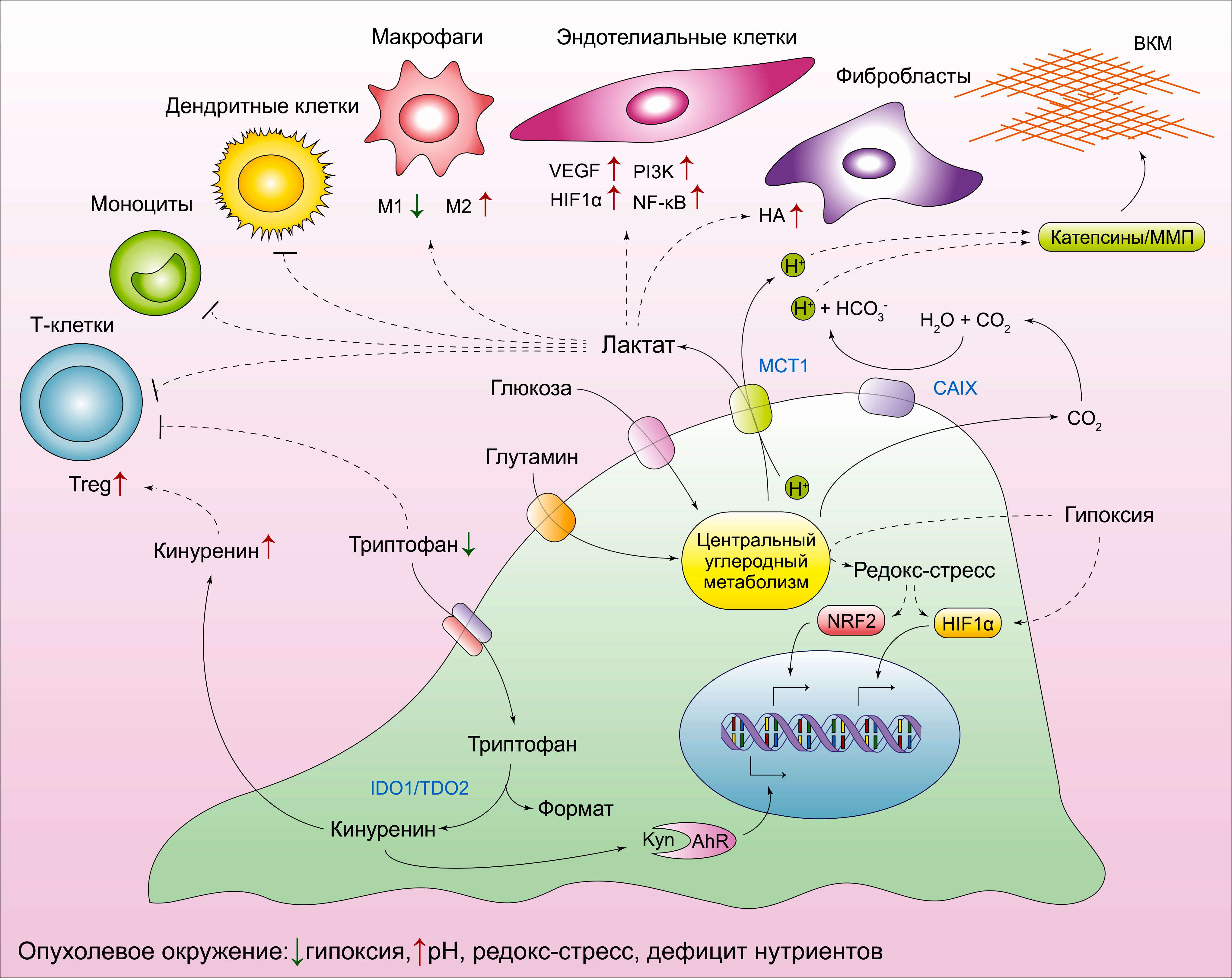
ECM, внеклеточный матрикс; Treg, регуляторные Т-клетки; HA, гиалуроновая кислота; MMPs, матриксные металлопротеиназы; MCT1, монокарбоксилатный переносчик 1; CAIX, карбоангидраза IX; IDO1, индоламин-2,3-диоксигеназа 1; TDO2, триптофан-2,3-диоксигеназа 2; Kyn, кинуренин; AhR, рецептор арильных углеводородов.
Секреция лактата во внеклеточное пространство через монокарбоксилатный транспортер MCT1 сочетается с ко-транспортом Н+, вызывающих закисление клеточного микроокружения. Помимо этого, избыток образованного в ходе митохондриальных реакций декарбоксилирования СО2 также ведёт к закислению внеклеточной среды. В конце концов, СО2 диффундирует во внеклеточное пространство, где преобразуется в H+ и HCO3- классом внеклеточных карбоангидраз. Экспрессия карбоангидраз, в частности изоформы CAIX, повышается во время гипоксии. Чрезмерное закисление внеклеточной среды стимулирует протеолитическую активность матриксных металлопротеиназ (ММП) и катепсинов, что ускоряет деградацию компонентов внеклеточного матрикса и способствует опухолевой инвазии.
Накопление лактата и соответствующая ей ацидификация внеклеточного пространства могут рассматриваться как побочные эффекты от опухоль-специфического метаболического перепрограммирования. Вместе с тем, некоторые опухоли применяют другую стратегию, состоящую в формировании вокруг себя малочувствительного к иммуному ответу микроокружения. Например, многочисленные типы солидных опухолей гиперэкспрессируют триптофан-деградирующие диоксигеназы, индоламин-2,3-диоксигеназу (ИДО1) и триптофан-2,3-диоксигеназу (ТДО2), катализирующие превращение незаменимой аминокислоты триптофана в его производное кинуренин. Более того, накопленный кинуренин выступает в качестве лиганда для арил-гидрокарбонового рецептора (АгР). Таким образом, действуя совместно с АгР, кинуренин способствует формированию фенотипа регуляторных Т-клеток, что в дальнейшем ведёт к подавлению противоопухолевого иммунного ответа. В конце концов, кинуренин потенцирует аутокринный сигнальный путь через АгР на сами опухолевые клетки, усиливая деградацию внеклеточного матрикса и инвазию. Малые молекулы-ингибиторы ИДО1 в настоящее время тестируются в рамках клинических испытаний.
С другой стороны, условия внутри опухолевого микроокружения имеют сильное влияние на метаболизм опухолевой клетки. Как было сказано ранее, опухоли зачастую сталкиваются с дефицитом питательных веществ и кислорода в окружающей их среде, что заставляет их создавать различные стратегии по преодолению этих ограничений. Более того, гипоксия блокирует проведение окислительного фосфорилирования и других зависимых от кислорода реакций в клетках, а также нарушает окислительно-восстановительный баланс, влияя, таким образом, на клеточные сигнальные пути и транскрипционную активность. В целом, взаимодействие между опухолевыми клетками и их микроокружением накладывает весьма ощутимый отпечаток на новообразование, формируя в дальнейшем метаболизм опухолевой клетки и активно способствуя появлению более агрессивных свойств.
Остальные вопросы
Метаболические перестройки, опосредованные онкогенами, позволяют опухолевым клеткам сохранять особенности своей нерегулируемой пролиферации, противостоять метаболическим проблемам, связанным с ограничениями в доступе к кислороду и питательным веществам, сохранять дедифференцированное состояние посредством повреждений в важнейших паттернах экспрессии генов, а также перестраивать окружающее микроокружение таким образом, чтобы оно активно способствовало опухолевому росту и диссеминированию. Пока большинство исследований к настоящему времени были сосредоточены на нарушениях метаболизма глюкозы и глутамина, опухолевые клетки используют и многие другие питательные вещества, среди которых можно выделить серосодержащие аминокислоты цистеин и метионин, незаменимые жирные кислоты, холин, металлы в следовых количествах и витамины. Мы только начинаем понимать степень необходимости этих нутриентов для опухолеобразования.
К примеру, существует множество доказательств, указывающих на то, что метаболизм серы может перестраиваться при канцерогенезе. Так, в опухолевых клетках повышается интенсивность захвата цистеина благодаря увеличению транскрипционной активности хСТ-транспортера, а также вторично за счёт наращивания темпов утилизации клеткой глутамина. Примечательно, что не менее 30% импортируемого в клетки глутамина, согласно исследованиям, покидает клетку через хСТ-транспортер в виде глутамата, с одновременным обменом на цистеин. По причине повышенной утилизации опухолевыми клетками, цистеин считается второй наиболее дефицитной аминокислотой в опухолях поджелудочной железы по сравнению с нормальной панкреатической тканью. Цистеин может подвергаться нескольким типам метаболических превращений в быстро пролиферирующих клетках, среди которых — биосинтез глутатиона и железосерных кластеров, а также сероводорода (H2S), выступающего газовым медиатором со сложными, не до конца определёнными функциями в физиологии клетки. К примеру, он принимает участие в защите от оксидативного стресса, избыточного митохондриального дыхания, блокирует апоптоз и способствует ангиогенезу.
На факты перестроек в опухолевом метаболизме серина стали обращать пристальное внимание лишь в течение последних 4 лет. Примечательно, что, помимо de novo синтезированного серина, на метаболизм одноуглеродных соединений и концентрацию SAM влияют также и вовлечение экзогенных доноров одноуглеродных групп. Кроме того, дефицит в алиментарно поступающих серине и глицине доказанно ослабляет рост ксенотрансплантированных клеток колоректального рака на экспериментальных моделях мышей. Среди других двух доноров метильных групп для образования SAM следует упомянуть такие алиментарно поступающие вещества, как холин и его производное бетаин. Повышение темпов захвата холина наблюдалось в клетках рака молочной железы и опухоли простаты; в связи с этим, радиоактивные молекулы 18F- и 11С-холина могут применяться в клинической ПЭТ-диагностике опухолей. Высокое алиментарное потребление холина связано с достоверно повышенным риском развития летального случая рака простаты. Напротив, диета с высоким содержанием холина и бетаина ассоциирована с пониженным риском возникновения рака молочной железы и рака лёгких. Более того, на многочисленных животных моделях было продемонстрировано, что холин- и бетаин-дефицитные диеты напрямую затрагивают процессы метилирования гистонов и ДНК. Это подразумевает, что нарушение в поступлении экзогенных метильных доноров может иметь значительный эффект на состоянии эпигенетической системы клетки и, таким образом, может вести к опухолеобразованию.
Тема эффекта от различных микронутриентов — таких как витаминов и металлических элементов в следовых количествах — на опухолевый рост рассматривается в качестве многообещающей сферы для исследований. Микронутриенты используются в качестве кофакторов для различных ферментов и играют сложную, до конца не определённую роль в регуляции клеточных метаболических процессов, а также в сигнальной трансдукции. К примеру, аскорбиновая кислота является кофактором α-кетоглутарат-зависимых диоксигеназ. Примечательно, что аскорбиновая кислота влияет на метилирование ДНК в эмбриональных стволовых клетках и фибробластах эмбрионов мыши, зависимых от ТЕТ2. В силу того, что многие злокачественные опухоли характеризуются повышенным метилированием островков СрG, нарушения в импорте аскорбиновой кислоты также может повлиять на их состояние эпигенетической системы. Более того, концентрации определённых металлических микроэлементов, таких как цинк и медь, дисрегулированы в опухолевых клетках, в сравнении с нормальными тканями. В частности, содержание меди может быть повышено в 2 или 3 раза в опухолях молочной железы и яичников, а также при лейкозах. Интересно, что медь, как недавно было выявлено, участвует в BRAFV600E-опосредованной активации сигнального пути ERK в клетках меланомы. Соответственно, конститутивная активация ERK-пути способна предупреждать эффект от истощения запасов меди.
Наконец, вклад широкого спектра самых разнообразных метаболитов, продуцируемого «позабытым всеми органом» тела — микробиотой — на инициацию опухолевого роста и прогрессии только начинает активно изучаться. В целом, концепция метаболических нарушений в опухолевых клетках, впервые замеченная около века назад, продолжает вскрывать новые связи между использованием нутриентов и состоянием опухолевого развития. Более того, исследование опухолевого метаболизма не только проливает свет на течение канцерогенеза, но и открывает новые принципы того, как регулируется биохимия пластического обмена и как она поддерживает рост, пролиферацию и дифференцировку нормальных клеток.
