
Дефицит лизосомной кислой липазы (болезнь Вольмана)
ДЛКЛ (LAL-D) — это гетерогенное заболевание, которое по разному проявляется и с разной скоростью прогрессирует у разных индивидов. Первый случай был описан в 1956 году у младенца. Несколько лет спустя Фредриксон сообщил о 12-летнем мальчике с выраженной гиперхолестеринемией, гепатомегалией и накоплением эфиров холестерола. Это состояние было названо болезнью накопления эфиров холестерола (БНЭХ, CESD — cholesteryl ester storage disease). Также было обнаружено, что болезнь Вольмана и БНЭХ имеют молекулярную патологию, возникающую в результате мутаций в гене LIPA, который кодирует лизосомную кислую липазу (ЛКЛ, LAL — lysosomal acid lipase) — фермент, ответственный за гидролиз эфиров холестерола и триглицеридов c частицами липопротеинов низкой плотности (ЛПНП, LDL — low density lipoprotein) в свободный холестерин и свободные жирные кислоты.
Генетика и наследование
ДЛКЛ возникает из-за мутаций в гене LIPA, который располагается в локусе 10q23,2 10 хромосомы, имеющий 10 экзонов, и его длина составляет приблизительно 45 кб; более 40 мутаций с потерей функции были обнаружены в этом гене. ДЛКЛ — аутосомно-рецессивное заболевание, самые серьезные изменения, такие как нонсенс мутации, сдвиг рамки считывания и точечные мутации приводят к стоп-кодонам, которые, как правило, обнаруживаются у младенцев. Менее серьезные мутации выявляются у детей постарше и взрослых. Чаще всего унаследованным дефектом является мутация в 8 экзоне, E8SJM (c.894G> A), которая имеется у половины всех детей и взрослых с ДЛКЛ. Мутация представляет собой альтернативный акцептор сайта сплайсинга, что в результате приводит к удалению 8 экзона в мРНК. Исследования в общей популяции показали, что частота аллеля E8SJM выглядит следующим образом: 0,0013 у представителей белой европеоидной расы (США — 0,0017, Германия — 0,0025, ЕС — 0,0012); 0,0017 у латиноамериканцев; 0,0010 у евреев Ашкенази; 0,0005 у азиатов и 0,0000 у афроамериканцев. Предполагаемая частота встречаемости в России 0,00001-0,00000667.
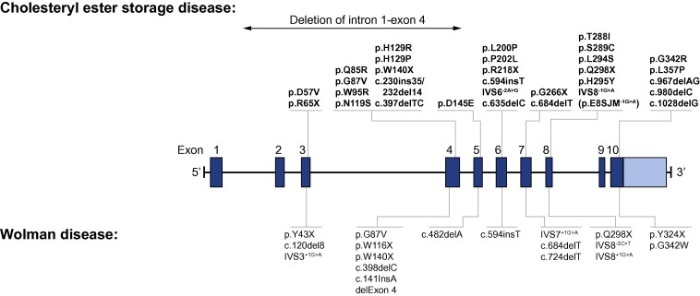
Рисунок 1 | Мутации в гене LIPA.
Патогенез
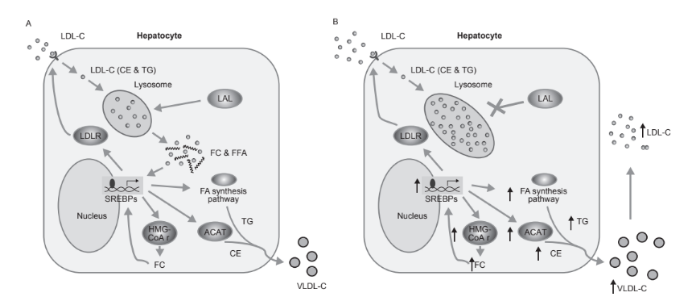
ЛКЛ играет ключевую роль в метаболизме липидов посредством гидролиза эфиров холестерола и триглицеридов в лизосомах. Полученные на основе ЛПНП нейтральные жиры (эфиры холестерола и, в меньшей степени, триглицериды) расщепляются с помощью ЛКЛ. Эти липиды или их окисленные производные взаимодействуют с факторами транскрипции SREBPs, которые непосредственно управляют экпрессией генов, участвующих в синтезе и поглощении холестерола, и липогенезе. Как правило, высокое внутриклеточное количество свободного холестерола приводит к SREBP-2-опосредованному снижению количества ЛПНП-рецепторов, в результате чего поступление холестерола в клетку уменьшается, ингибированию по типу обратной связи гидроксиметилглутарил-кофермента A редуктазы (ГМГ-КоА-редуктаза), приводящее к снижению синтеза холестерола, и стимуляции ацил-холестерола ацилтрансферазы, который ускоряет образование сложных эфиров холестерола.
Когда активность ЛКЛ снижается или исчезает, сложные эфиры холестерола и триглицериды не расщепляются и накапливаются в лизосомах. Следовательно, дефицит внутриклеточного свободного холестерола вызывает SREBP-опосредованную активацию синтеза эндогенного холестерола посредством ГМГ-КоА-редуктазы и эндоцитоз через ЛПНП-рецепторы, а также повышение синтеза аполипопротеина B (ApoB) и заметное увеличение синтеза липопротеинов очень низкой плотности.
Клиническая картина
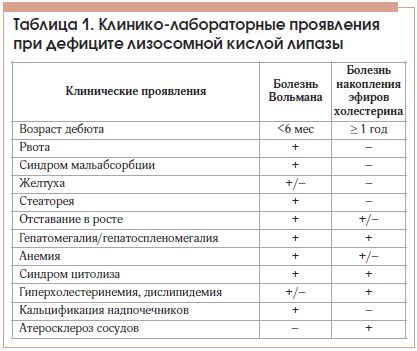
Многие из наиболее распространенных клинических проявлений, а именно дислипидемия, гепатомегалия и повреждение клеток печени (о чем свидетельствует увеличение сывороточных трансаминаз с прогрессированием, которое ведет к фиброзу и циррозу) дифференцируют с другими сердечно-сосудистыми заболеваниями, болезнями печени и метаболическими отклонениями, распространенными больше, чем ДЛКЛ. Младенцы с ДЛКЛ обычно имеют более острое течение болезни по сравнению с детьми и взрослыми. Желудочно-кишечные симптомы (рвота, диарея со стеатореей и вздутие живота) и нарушение роста часто являются первыми проявлениями. Точечный кальциноз надпочечников может наблюдаться на рентген-снимке примерно у 50% младенцев и может также наблюдаться у детей. Анемия также может появляться у данных больных.
Источники
- Reiner Ž. et al. Lysosomal acid lipase deficiency–an under-recognized cause of dyslipidaemia and liver dysfunction //Atherosclerosis. – 2014. – Т. 235. – №. 1. – С. 21-30.
- Pisciotta L. et al. Molecular and clinical characterization of a series of patients with childhood-onset lysosomal acid lipase deficiency. Retrospective investigations, follow-up and detection of two novel LIPA pathogenic variants //Atherosclerosis. – 2017. – Т. 265. – С. 124-132.
- Strokova Т.V., Bagaeva M.E., Matinyan I.A. Lysosomal acid lipase deficiency // RMJ. 2017. № 19. P. 1346–1351.
