
Современные представления о хронической гранулематозной болезни (ХГБ)
Хроническая гранулематозная болезнь (ХГБ) — достаточно неплохо изученное на сегодняшний день наследственное заболевание, связанное с недостаточностью фагоцитарной системы, следствием которой является иммунодефицит. Впервые ХГБ была описана в 1959 году у детей, для которых болезнь оказалась смертельной [1]. Болезнь не поддавалась лечению; на аутопсии выявляли генерализованное гранулематозное воспаление, отличающееся по своей специфике от известных на тот момент инфекционных и неинфекционных заболеваний.
Сегодня нам известно намного больше. Заболевание в большинстве случаев более не является смертельным, выявляется в основном у детей (врожденный иммунодефицит проявляется достаточно быстро), неплохо купируется. Однако до сих пор ХГБ относится к заболеваниям неизлечимым.
Этиология и патогенез
Сущность хронической гранулематозной болезни — в недостаточности фермента НАДФ-оксидазы, который представляет собой комплекс каталитических протеинов. Этот фермент, находясь в фагоцитирующих клетках, обеспечивает перенос электрона с НАДФ на молекулярный кислород: происходит т. н. «респираторный взрыв» (см. Рис. 1). Строго говоря, данный фермент есть не только у фагоцитов: единственным специфичным для них белком является трансмембранный gp91, остальные же компоненты встречаются в самых разных клетках [2].
Именно этот респираторный взрыв и позволяет завершить фагоцитоз и лизировать микроб, в противном же случае будет наблюдаться феномен эндоцитобиоза — он же «незавершенный фагоцитоз» — когда микроб просто живет внутри клетки. Фагоцит вполне себе поглотит бактерию, а убить не сможет. Таков механизм развития многих тяжелых инфекций (например, туберкулеза) [3]; эта же патология лежит в основе и других первичных иммунодефицитов [4].
Обратимся к рисунку 1. Белки gp91 и p22 объединяют в цитохром b558 — мембраносвязанную часть фермента НАДФ-оксидазы, остальные протеины называют цитозольными. При активации фагоцита различными медиаторами цитозольные p47 и p67 фосфорилируются и связываются вместе. Комплекс приобретает сродство к белкам p47 и rac2 — таким образом, присоединяясь к ним, данные протеины вызывают конформационные изменения в мембранном цитохроме b558, — и комплекс приобретает оксидазную активность [2, 5]. НАДФ-оксидаза переносит электрон от своего кофермента НАДФ на кислород с формированием активных форм кислорода (АФК) — O2- и H2O2. Вот здесь и начинается самое интересное.
Классически считается, что фагоцит убивает микробы, образуя фаголизосому с бактерией или грибом, воздействуя на них большими дозами высокотоксичных АФК; однако в последнее время приобретает актуальность иная точка зрения. В 2002 году в Nature была опубликована статья, авторы которой пересмотрели всю парадигму деактивации поглощенных микробов.
Ученые обнаружили, что если в фагоцитах мышей определяется нормальный уровень активных форм кислорода, но имеется недостаточность лизосомальных ферментов — животные будут беззащитны против стафилококковых и кандидозных инфекций. То есть, несмотря на наличие нормального респираторного взрыва, иммунодефицит все равно присутствует. Стало быть, эффекторами в инактивации микроба являются не сами АФК [6].
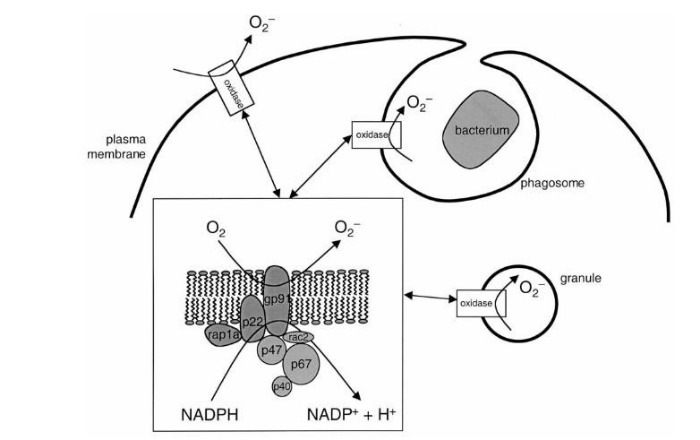 Рисунок 1. Механизм формирования NADPH-оксидазы [5].
Рисунок 1. Механизм формирования NADPH-оксидазы [5].
Согласно новой модели, АФК являются медиаторами в уничтожении микроба, а непосредственные «убийцы» — ферменты лизосом, обладающие протеолитической активностью. Как известно, при захвате фагоцитом микроба формируется вакуоль — (фагосома), с которой впоследствии сливается лизосома макрофага [7]. На мембране этой вакуоли и функционирует НАДФ-оксидаза, постоянно увеличивая концентрацию АФК. Однако как уже было сказано, данный процесс — не конечный эффекторный механизм.
Предположительно, супероксид-анион (O2-) вызывает приток ионов калия. K+, в свою очередь, приводит pH к оптимальным для функционирования протеолитических ферментов показателей. В роли калиевых каналов, возможно, может выступать как обычный протонный канал, так и сам комплекс НАДФ-оксидазы (который в данном случае будет представлен в качестве белка-переносчика) [6, 8].
Наследование и генетика
Хроническая гранулематозная болезнь имеет наследственную природу. Приблизительно данной патологией страдает 1 на 250 000 [9], что делает заболевание достаточно редким и потому трудным в диагностическом отношении. Заболевание вызывает мутация любого из четырех генов, кодирующих субъединицы ключевого фермента фагоцитоза НАДФ-оксидазы. Более двух третей случаев связаны с X-сцепленным наследованием (дефект гена CYBB, кодирующего белок p-91); остальные случаи связаны с аутосомно-рецессивным наследованием генов CYBA, NCF-1 и NCF-2, кодирующих белки p22, p47 и p67 соответственно. Исходя из этого болезнь обозначают как ХГБ X91, A22, A47 и A67 (в зависимости от типа наследования и локуса гена) [10].
Примечательно, что в литературе нет (или крайне мало) доказанных случаев наследственного дефекта других субъединиц. Однако в последнее время обнаруживаются все новые мутации генов, следствием которых становится ХГБ: например, в 2009 году выделили еще один подвид ХГБ, связанный с аутосомно-рецессивной мутацией гена p-40 [11], имеются также сведения о единственном пациенте с недостаточностью белка Rac2 — [10]. Вполне вероятно, что могут существовать и другие генетические патологии, вызывающие данное заболевание.
Диагностика
Диагностика ХГБ основана на выявлении клинических признаков, кроме того, важно выявить наличие или отсутствие респираторного взрыва. Последнее можно осуществить несколькими гистологическими и иммунологическими методами, например, окрашивание нитросиним тетразолием (НТЗ) позволяет определить, вырабатывают ли клетки АФК — НТЗ будет утилизироваться активными формами кислорода, в результате чего из бледно-желтого тетразолия образуется голубой формазан [12].
Среди наиболее ярких клинических симптомов можно выделить: пиодермию, пневмонию, воспалительные процессы желудочно-кишечного тракта, лимфаденит, абсцесс печени и остеомиелит [13] на фоне рецидивирующих бактериальных и грибных инфекций.
В крови выявляется гипергаммаглобулинемия и анемия. В местах дренажей — хроническое воспаление с образованием гранулем. Также гранулемы могут формироваться в различных тканях и органах: например, в желудке гранулематозное воспаление способно привести к обструкции желудочного канала, в урогенитальном тракте — к циститу. Также следует упомянуть, что почти 20 % больных ХГБ страдает от гранулематозного колита, который легко перепутать с болезнью Крона [14].
Основную же опасность для жизни пациента представляют различные инфекции. Примечательно, что поражают организм достаточно небольшой спектр бактерий и грибов: Staphylococcus, Burkholderia, Serratia, Nocardia, и некоторые грибы рода Aspergillus, реже — Salmonella и Mycobacterium tuberculosis [15, 16]. Несколько реже встречается инфицирование Chromobacterium violaceum (бацилла семейства Neisseriaceae, вызывающая тяжелые инфекционные осложнения в виде кожных фурункулов, абсцессов внутренних органов и септического поражения) [17]. Все они требуют специфического лечения. Кроме того, поражение органов также может быть стереотипным (см. Рис.2).
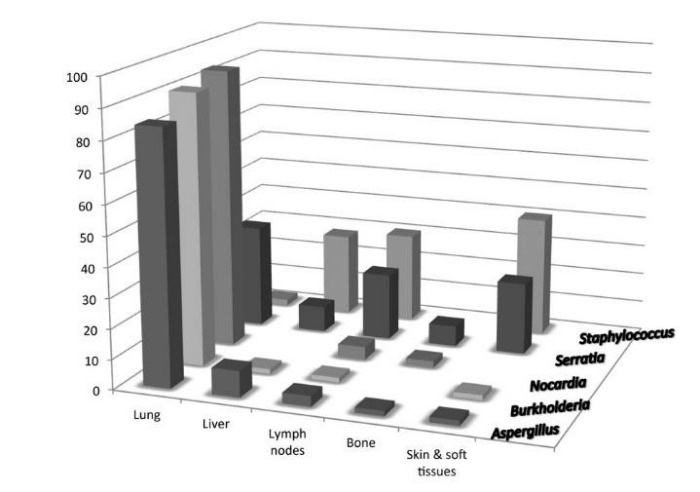 Рисунок 2. Сравнительная характеристика частоты органных поражений при некоторых инфекциях у пациентов с ХГБ [15].
Рисунок 2. Сравнительная характеристика частоты органных поражений при некоторых инфекциях у пациентов с ХГБ [15].
Также следует упомянуть о недавно открытой бактерии, выявленной у пациентов с ХГБ, которую исследователи предложили назвать Granulobacter bethesdensis. Это грамотрицательная палочка, которая на данный момент не может быть отнесена ни к одной из существующих таксономических групп; бактерия поражает лимфоузлы, кроме того, резистентна к антибиотикам in vitro и, скорее всего, — in vivo [18, 19]. Предполагается, что это — лишь первая из подобных бактерий, возникшая в эру антибиотиков. На данный момент Granulobacter bethesdensis не слишком распространена, однако имеет к этому весьма опасную тенденцию.
Лечение
В настоящее время активно разрабатываются методики, которые смогут не просто позволить больным с ХГБ жить полноценно, но и в перспективе совершенно избавить их от бремени заболевания. Поскольку болезнь наследственная, крайне трудно придумать что-то существенное, однако такие попытки предпринимаются, и некоторые из них обнадеживают.
Одна из них — лечение хронической гранулематозной болезни с помощью генной инженерии [20]. В 2006 году в Nature Medicine была опубликована статья, авторы которой сообщили об успешной коррекции генома двух пациентов с X-сцепленной формой ХГБ. После лечения у пациентов определяли активность нейтрофилов с помощью позитронно-эмиссионной томографии, а также других инструментальных методов. Исследование показало, что в обоих случаях фагоциты после проведенного лечения смогли оказать сопротивление инфекции. На данный момент это — одна из самых многообещающих методик.
Еще одним способом терапии является пересадка гемопоэтических клеток [21]. Двадцати семи пациентам после миелоаблативного режима кондиционирования (подготовка пациента к трансплантации с помощью лучевой или цитостатической терапии — прим. автора) пересадили гемопоэтические стволовые клетки от наиболее подходящих доноров (по белкам HLA — главного комплекса гистосовместимости). Двадцать три пациента вполне успешно перенесли операцию, а дальнейшее наблюдение позволило говорить об излечении этих пациентов от хронической гранулематозной болезни. Однако еще 4 пациента умерли от последующих инфекций (15 %).
В остальном же современная медицина может предложить крайне немного. Это — патогенетическая и симптоматическая терапия с использованием антибиотиков, дренажей и прочего. Излишне говорить, что подобное лечение не способно избавлять пациентов от ХГБ.
Профилактика инфекций.
Крайне важно предотвращение развития инфекционного процесса. Пациенты с ХГБ испытывают невероятные трудности в повседневной жизни, например, некачественная чистка зубов спокойно может окончиться гингивитом, а царапина — тяжелой бактериемией. Потому больным необходимо тщательным образом следить за гигиеной, выполнять профессиональную чистку зубов, обрабатывать все царапины антисептиком. И, разумеется, такие пациенты должны быть привиты по всем правилам [14]. В противном же случае даже достаточно простая инфекция может окончиться летально.
Источники:
- R. A. BRIDGES, H. BERENDES, and R. A. GOOD, “A Fatal Granulomatous Disease of Childhood,” AMA. J. Dis. Child., vol. 97, no. 4, p. 387, 1959.
- S. M. Holland, “Chronic Granulomatous Disease,” Clin. Rev Allerg Immunol, no. June 2009, pp. 3–10, 2010.
- B. D. L. Clemens and M. a Horwitz, “Characterization of the Mycobacterium mberctdosis Phagosome and Evidence that Phagosomal Maturation Is Inhibited,” Culture, vol. 181, no. January, 1995.
- W. Al-Herz et al., “Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency,” Front. Immunol., vol. 2, no. November, pp. 1–26, 2011.
- C. Dahlgren and A. Karlsson, “Respiratory burst in human neutrophils Claes,” J. Immunol. Methods, vol. 232, pp. 3–14, 1999.
- E. P. Reeves et al., “Killing activity of neutrophils is mediated through activation of proteases by K+flux,” Nature, vol. 416, no. 6878, pp. 291–297, 2002.
- A. Aderem and D. M. Underhill, “Mechanisms of Phagocytosis in Macrophages,” Annu. Rev. Immunol., vol. 17, no. 1, pp. 593–623, 1999.
- A. Maturana et al., “Heme Histidine Ligands within gp91phox Modulate Proton Conduction by the Phagocyte НАДФ Oxidase,” J. Biol. Chem., vol. 276, no. 32, pp. 30277–30284, 2001.
- J. M. van den Berg et al., “Chronic granulomatous disease: The European experience,” PLoS One, vol. 4, no. 4, pp. 1–10, 2009.
- P. G. H. Ãy, A. R. Cross, and J. T. C. Ã, “Chronic granulomatous disease,” no. 15, pp. 578–584, 2003.
- J. D. Matute et al., “A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil НАДФ oxidase activity,” Blood, vol. 114, no. 15, pp. 3309–3316, 2009.
- M. D. . Brahm H. Segal, P. D. . Thomas L. Leto, M. D. . John I. Gallin, M. D. . Harry L. Malech, and M. D. Steven M. Holland, “Genetic, biochemical, and clinical features of chronic granulomatous disease.” 2000.
- J. A. Winkelstein et al., “Chronic Granulomatous Disease.” 2000.
- R. A. Seger, “Modern management of chronic granulomatous disease,” Br. J. Haematol., vol. 140, no. 3, pp. 255–266, 2008.
- B. E. Marciano et al., “Common severe infections in chronic granulomatous disease,” Clin. Infect. Dis., vol. 60, no. 8, pp. 1176–1183, 2015.
- N. Bennett, P. J. Maglione, B. L. Wright, and C. Zerbe, “Infectious Complications in Patients With Chronic Granulomatous Disease,” vol. 7, no. June, 2018.
- Z. Meher-Homji, R. P. Mangalore, P. D. R. Johnson, and K. Y. L. Chua, “Chromobacterium violaceum infection in chronic granulomatous disease: a case report and review of the literature,” JMM Case Reports, vol. 4, no. 1, 2017.
- D. E. Greenberg et al., “A novel bacterium associated with lymphadenitis in a patient with chronic granulomatous disease,” PLoS Pathog., vol. 2, no. 4, pp. 260–267, 2006.
- D. E. Greenberg et al., “Recurrent granulibacter bethesdensis infections and chronic granulomatous disease,” Emerg. Infect. Dis., vol. 16, no. 9, pp. 1341–1348, 2010.
- M. G. Ott et al., “Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1,” Nat. Med., vol. 12, no. 4, pp. 401–409, 2006.
- R. A. Seger et al., “Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: A survey of the European experience, 1985-2000,” Blood, vol. 100, no. 13, pp. 4344–4350, 2002.
