
«Свой среди чужих, чужой среди своих» — некроптоз
Некроптоз называют «запрограммированной формой некроза», так как он демонстрирует морфологические признаки, сходные с некрозом (набухание клеток с последующим разрывом плазматической мембраны), но при этом имеет четкую систему регуляции. Впервые был открыт в 1996 году в гепатоцитах свиньи, инфицированных вирусом коровьей оспы, который экспрессирует модификатор цитокиновой реакции A (CrmA), а также вирусный ингибитор CASP1 и CASP8 (что делает апоптоз данных условиях невозможным) [1]. Предполагается, что эволюционное значение этой формы клеточной смерти состоит в обеспечении резервного пути элиминации клеток в случаях ингибирования апоптотического пути.
Некроптоз запускают различные стимулы, включая активацию рецепторов смерти (ключевую роль играет TNFRSF1A из надсемейства рецепторов фактора некроза опухоли, активирующий транскрипционный фактор «каппа-би» NF-κB; также возможна активация через FAS-рецептор), Toll-подобных рецепторов (ТLR3 и TLR4), внутриклеточных сенсоров нуклеиновых кислот (например, Z-ДНК-связывающий белок 1 (ZBP1), также известный как DAI), рецепторов ретиноевой кислоты протеин 3 респондеров (RARRES3 или RIG1), белка эндоплазматического ретикулума, стимулятора генов интерферонов 173 (TMEM173 или STING) и рецепторов адгезии. Лиганды, активирующие внешний путь апоптоза (например, TNFSF10 или TRAIL, Fas-лиганд FASLG/FasL/CD95L), также могут вызывать некроптоз при блокировке действия CASP8 и вызывающего смерть сигнального комплекса (DISC) ингибиторами каспаз (Z-VAD-FMK) или в случае истощения Fas-ассоциированных доменов смерти (FADD) [2].
Некроптоз не только опосредует адаптивный ответ при стресс-индуцированном нерепарируемом повреждении клеток, но также участвует в программах защиты эмбрионального развития (устранение потенциально дефектных организмов внутриутробно), а также в поддержании гомеостаза Т-клеток у взрослых [3].
На молекулярном уровне некроптоз зависит от последовательной активации рецептора протеинкиназы-3 (RIPK3) и киназного домена смешанной линии, подобного псевдокиназе (MLKL) (рис.1). После инициации некроптоза RIPK3 активируется RIPK1 (при условии, что CASP8 неактивна — отличие от апоптоза) посредством взаимодействия между гомотипическими RHIM-доменами. Фосфорилирование этих белков в их киназном домене способствует RHIM-опосредованному взаимодействию, что приводит к образованию амилоидоподобного нитевидного сигнального комплекса, называемого некросомой.
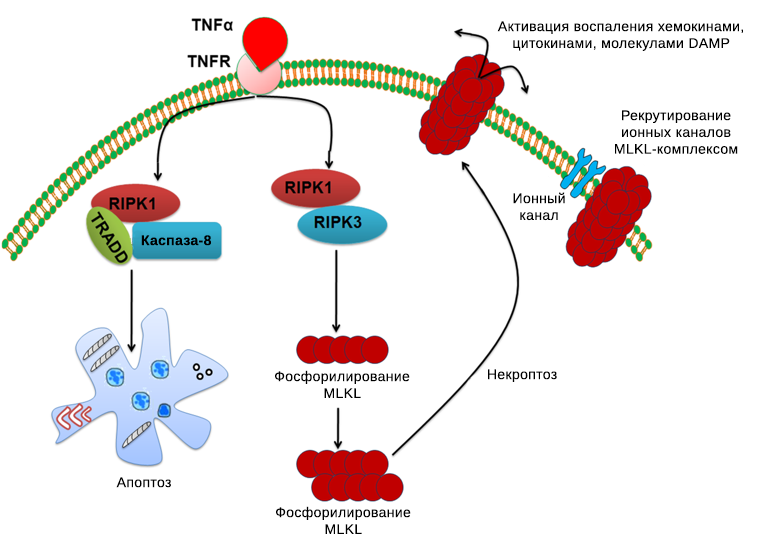
Рисунок 1 | Молекулярные механизмы апоптоза и некроптоза. TRADD — адаптер для рецептора фактора некроза опухоли 1 типа (англ. TNFR1-associated DD-protein — белок, взаимодействующий с доменом смерти рецептора TNFR1); RIPK1 и RIPK3 — рецепторы серин-треониновых протеинкиназ 1 и 3; TNFα и TNFR — фактор некроза опухоли α и его рецептор.
Рецепторы смерти опосредуют как внешний путь апоптоза, так и некроптоз. Активация каспазы-8 запускает апоптоз, в то время как ее ингибирование приводит к некроптозу. Взаимодействие RIPK1 и RIPK3 приводит к образованию функционального гетеродимерного комплекса, способствующего олигомеризации MLKL путем его фосфорилирования. Олигомерная форма MLKL транслоцируется из цитозоля в направлении цитоплазматической мембраны, что приводит к образованию поры, запуская воспалительный ответ. MLKL также осуществляет свою функцию, взаимодействуя с ионными каналами [4].
Интересно, что другие белки, такие как TLR3/4, TRIF и DAI (ДНК-активатор интерферона), также имеют домен RHIM, следовательно, они также могут образовывать некросомы, которые считаются неклассическими.
Также RIPK3 может быть активирован после RHIM-зависимого взаимодействия с TRIF, либо при активации TLR3 двухцепочечной РНК в эндосомах, либо при активации TLR4 липополисахаридом (LPS) или различными DAMP на плазматической мембране, или ZBP1, который действует как сенсор для синтеза цитозольного интерферона I типа, стимулирующего ДНК. Эти молекулы так же, как и активация рецептора фактора некроза опухоли (TNFRSF1A), способны запустить сигнальный путь NF-κB.
Активный RIPK3 катализирует фосфорилирование MLKL, что приводит к образованию олигомеров MLKL (наиболее вероятно, тримеров или тетрамеров), которые транслоцируются в цитоплазматическую мембрану. Далее MLKL действует двумя способами:
1. Как «платформа» в плазматической мембране для рекрутирования ионных каналов Na+ или Ca2+;
2. Способствует образованию пор в цитоплазматической мембране, взаимодействуя с амино-концом фосфатидилинозитолфосфата [4].
В случае апоптоза секреция цитокинов отсутствует или очень мала, тогда как во время некроптоза это ключевой этап, приводящий к выраженному воспалению. Потеря целостности клетки вызывает мощный провоспалительный эффект, связанный с выбросом эндогенных (в физиологическом состоянии локализующихся внутриклеточно) молекулярных паттернов опасности (danger assotiated molecular patterns, DAMP).
Логично предположить, что химические ингибиторы RIPK1 (такие как некростатин-1 (Nec-1) и его производные, например, Nec-1s) надежно ингибируют некроптоз in vitro и in vivo. Выключение этого механизма гибели клеток рассматривается как терапевтическая мишень при нейродегенеративных заболеваниях (табл. 1).
Таблица 1 | Роль некростатина-1 (Nec-1) в терапии нейродегенеративных заболеваний

Эффективность Nec-1 продемонстрирована и при ряде других заболеваний, включая ишемический инфаркт головного мозга, инфаркт миокарда, TNF-индуцированный синдром системного воспалительного ответа (SIRS), конканавалин А- и ацетаминофен-индуцированные гепатиты, ишемию/реперфузионное повреждение почек (ИРП) [8].
Некроптоз способствует эмбриональному и постнатальному развитию, участвует в поддержании гомеостаза тканей, а также в развитии различных патологических состояний. Ключевые молекулы некроптоза RIPK3 и RIPK1 могут стать новыми мишенями при лечении нейродегенеративных заболеваний и множества других, в патогенезе которых участвует воспалительный компонент.
Источники:
1. Berghe T.V., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47.
2. Moriwaki K., Chan F.K.M. Regulation of RIPK3-and RHIM-dependent necroptosis by the proteasome. J Biol Chem. 2016;291:5948–59.
3 Newton Kю, Wickliffe K.E., Maltzman A., Dugger D.L., Strasser A., Pham V.C., et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33.
4. Dhuriya Y. K., Sharma D. Necroptosis: a regulated inflammatory mode of cell death. Journal of Neuroinflammation.2018;15:199 .
5. Yang S.H., Lee D.K., Shin J., Lee S., Baek S., Kim J., et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med. 2017;9:61–77.
6. Iannielli A., Bido S., Folladori L., Segnali A., Cancellieri C., Maresca A. et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson's Disease Models. Cell Rep. 2018;22:2066–79.
7. Zhu S., Zhang Y., Bai G., Li H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell death & disease. 2011; 2(1), e115.
8. Conrad M., Angeli J. P. F., Vandenabeele P., Stockwell B. R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery. 2016;15: 348–366.
