
Мутагенная репликация: возможная мишень для терапии опухолей

Исследование, опубликованное в Cell, доказывает возможность повышения чувствительности раковых клеток к химиотерапии, повреждающей ДНК, путем медикаментозного ингибирования синтеза мутагенной ДНК на поврежденной матрице.
Классическая генотоксическая химиотерапия рака направлена на уничтожение пролиферирующих опухолевых клеток путем повреждения ДНК, за счет чего останавливается репликация. Запускается реакция повреждения ДНК, приводящая к остановке клеточного цикла, старению или апоптозу. Химиотерапевтические препараты используются для лечения рака, однако повреждают не только раковые клетки, но и здоровые, что вызывает побочные эффекты: выпадение волос, ослабление здоровья желудочно-кишечного тракта и кроветворной системы и в долгосрочной перспективе преждевременное старение. Кроме того, химиотерапевтические препараты — мутагены, поэтому способны ускорить прогрессирование опухоли и даже спровоцировать появление вторичных опухолей (рис. 1).
.
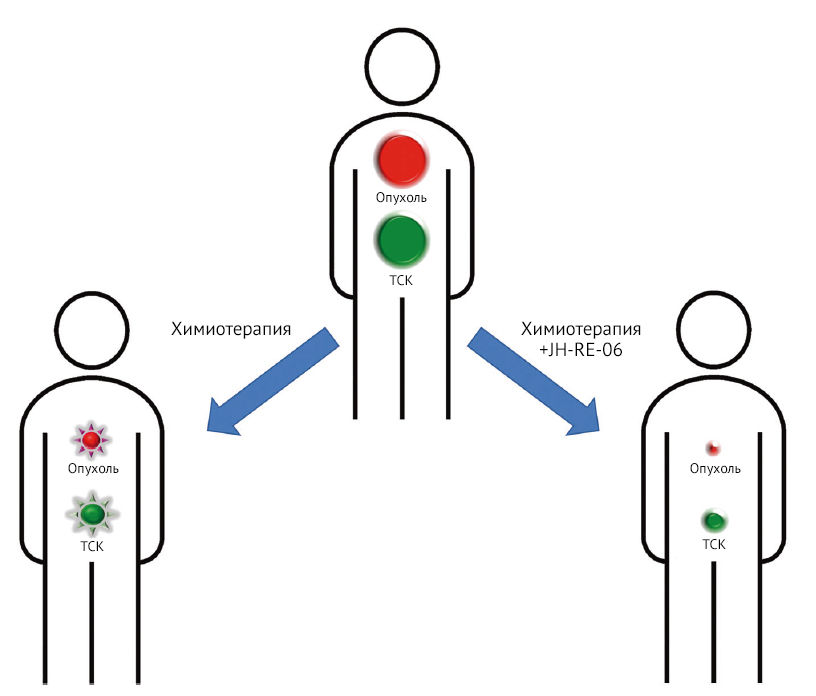
звезда) или вторичному онкогенезу (зеленая звезда). JH-RE-06 ингибирует опосредованный REV1 мутагенный транслезионный синтез. Это приводит к сенсибилизации пролиферирующих опухолевых клеток (и, возможно, также нормальных стволовых клеток) к токсическим эффектам химиотерапии, при этом мутагенные последствия транслезионного синтеза не развиваются.
Тем не менее нельзя исключить того, что системная инактивация REV1 может спровоцировать нежелательные мутации.
Цитотоксические повреждения ДНК вследствие патологических изменений отдельных нуклеотидов подавляются с помощью механизмов устойчивости. Синтез через поврежденные участки — транслезионный синтез — основной механизм устойчивости к повреждениям, действует за счет ДНК-полимераз, которые реплицируются через измененные нуклеотиды. Это снижает репликационный стресс и подавляет реакцию на повреждение ДНК.
Транслезионные полимеразы реплицируют поврежденные нуклеотиды и не только способствуют толерантности к изменениям нуклеотидов, но и генерируют мутации, которые связаны с инициацией и прогрессированием опухоли. Возникла идея, что ингибирование мутагенного транслезионного синтеза повысит чувствительность рака к генотоксической химиотерапии, а также позволит снизить ее мутагенность и онкогенность (рис. 1).
Чтобы изучить новый подход к химиотерапии рака, Wojtaszek и соавторы провели скрининг низкомолекулярных ингибиторов REV1 — белка, который, помимо транслезионного синтеза в участках с удаленными азотистыми основаниями и поражениями нуклеотидов в малой бороздке спирали, контролирует другие транслезионные полимеразы, включая полимеразу η и REV7, субъединицу транслезионной полимеразы zeta. Из 10000 соединений ученые идентифицировали производное 1,4-дигидрохинолин-4-она — JH-RE-06, которое индуцирует ассиметричную димеризацию молекул белка REV1 в С-концевых доменах. Это блокирует домен REV1, связывающий REV7, и, следовательно, блокирует REV1/REV3/REV7-опосредованный мутагенный транслезионный синтез.
В экспериментах с использованием фибробластов эмбрионов мыши с удаленным REV1 или человеческих клеток с низким уровнем REV1 было доказано, что JH-RE-06 специфически ингибирует опосредованный REV1 путь мутагенного транслезионного синтеза при поражениях нуклеотидов, индуцированных химиотерапевтическими препаратами. После введения химиотерапевтического препарата цисплатина ученые подавили транслезионный синтез через сайт-специфическое поражение ДНК при помощи JH-RE-06. При обработке фибробластов мышиных эмбрионов комбинацией цисплатина и JH-RE-06 отмечалось снижение частоты мутаций до уровня ниже частоты «спонтанных» мутаций, то есть наблюдаемых в отсутствие лечения цисплатином. JH-RE-06 увеличивал цитотоксичность цисплатина и других агентов, повреждающих ДНК, по отношению к линиям раковых клеток и иммортализованным фибробластам мышиных эмбрионов дикого типа, но не по отношению к первичным фибробластам человека.
Затем авторы исследовали свойства JH-RE-06 как стимулятора сенсибилизации раковых клеток к химиотерапии. С этой целью ученые вводили цисплатин, JH-RE-06 или JH-RE-06 и цисплатин одновременно непосредственно в клетки ксенотрансплантатов меланомы мышей. По сравнению с опухолями, которые обрабатывали одним компонентом, комбинированная обработка подавляла рост опухоли в большей степени, и такие мыши жили на 50 % дольше. Таким образом, авторы продемонстрировали, что низкомолекулярный ингибитор REV1 повышает чувствительность раковых клеток к цисплатину как in vitro, так и in vivo, подавляя индуцированный мутагенез in vitro. Подавление мутагенеза с помощью генотоксической химиотерапии после ингибирования транслезионного синтеза способно замедлять развитие опухоли и предотвращать рецидивы.
Связь между транслезионным синтезом и раком сложнее, чем предполагалось. Как ни парадоксально, нарушение транслезионного синтеза способно ускорять канцерогенез, а не наоборот. Кроме того, мутагенный транслезионный синтез необходим для поддержания нормальных тканей, обеспечивая толерантность не только к генотоксическим препаратам, но также к эндогенным нуклеотидным поражениям. Транслезионный синтез играет значимую роль в диверсификации иммунной системы. Системное воздействие ингибиторов REV1 способно усиливать эндогенную или индуцированную химиотерапией токсичность по отношению к пролиферирующим нормальным тканям.
Работа ученых доказывает, что ингибирование REV1 улучшает результаты химиотерапии рака. Важно изучить возможные краткосрочные и долгосрочные последствия системного ингибирования REV1, прежде чем применять методику в клинике.
