
Трансформация эндотелиальных клеток при атеросклерозе

Клетки, способствующие развитию атеросклероза, очень пластичны и могут менять свой фенотип при изменении микроокружения. Исследование в Nature Metabolism показало, что трансформирующий фактор роста-β (ТФР-β) может превращать эндотелиальные клетки в провоспалительные, а ингибирование передачи сигналов от ТФР-β-рецептора к эндотелию может повернуть вспять развитие атеросклероза у мышей.
Эндотелиальные клетки (ЭК) выстилают внутреннюю поверхность кровеносных сосудов и являются важными участниками воспалительных реакций. В ходе развития атеросклероза ЭК подвергаются хроническому воспалению из-за сочетания турбулентного кровотока, накопления липидов в стенке сосуда и воздействия воспалительных медиаторов (например, ИЛ-1β). Активированный воспалительным процессом эндотелий обеспечивает сохранение медиаторов воспаления в образующейся атеросклеротической бляшке. Понимание того, как именно ЭК реагируют на воспалительное микроокружение, прогрессирование воспалительного процесса и поддержание хронического воспаления в бляшке необходимо для большей эффективности терапии.
Недавние клинические испытания показали, что при полном блоке передачи сигналов ИЛ-1β уменьшались сердечно-сосудистые события у пациентов с хроническим системным воспалением, таким образом обеспечив доказательство того, что воздействие на воспалительный процесс может быть новым подходом в борьбе с атеросклерозом. Тем не менее, антагонизм к ИЛ-1β также связан с повышенным риском развития инфекций, поэтому оправдано применение таргетной терапии. Изучение реакции эндотелия на воспалительные стимулы не является единственной целью. Помимо экспрессии молекул адгезии лейкоцитов и производства хемокинов, которые связываются с циркулирующими резидентными и воспалительными клетками, активированные ЭК могут превращаться в мезенхимоподобные клетки в процессе, известном как эндотелиально-мезенхимальный переход (EndMT).
Эти клетки либо коэкспрессируют ЭК и мезенхимальные маркеры (стадия частичного эндотелиально-мезенхимального перехода), либо полностью теряют идентичность; ЭК отслаиваются от эндотелия и входят в популяцию фибробластов или (в меньшей степени) в группу гладкомышечных клеток внутри бляшки. Так как EndMT связан с определенными клиническими событиями, понимание регулирования этого процесса может привести к разработке принципиально новых терапевтических подходов.
Цитокин ТФР-β недавно был признан проатеросклеротическим, ключевым фактором EndMT (рис. 1). Однако роль передачи сигналов ТФР-β в атерогенезе противоречива, т. к. исследования на людях показали причастность ТФР-β к стабилизации бляшки. Данное противоречие отражает различные роли лигандов, рецепторов и факторов транскрипции в этом процессе. Хотя глобальное ингибирование передачи сигналов ТФР-β предполагает, что ТФР-β подавляет атеросклероз in vivo, вклад ТФР-β сигналинга непосредственно в эндотелии не тестировался.
.
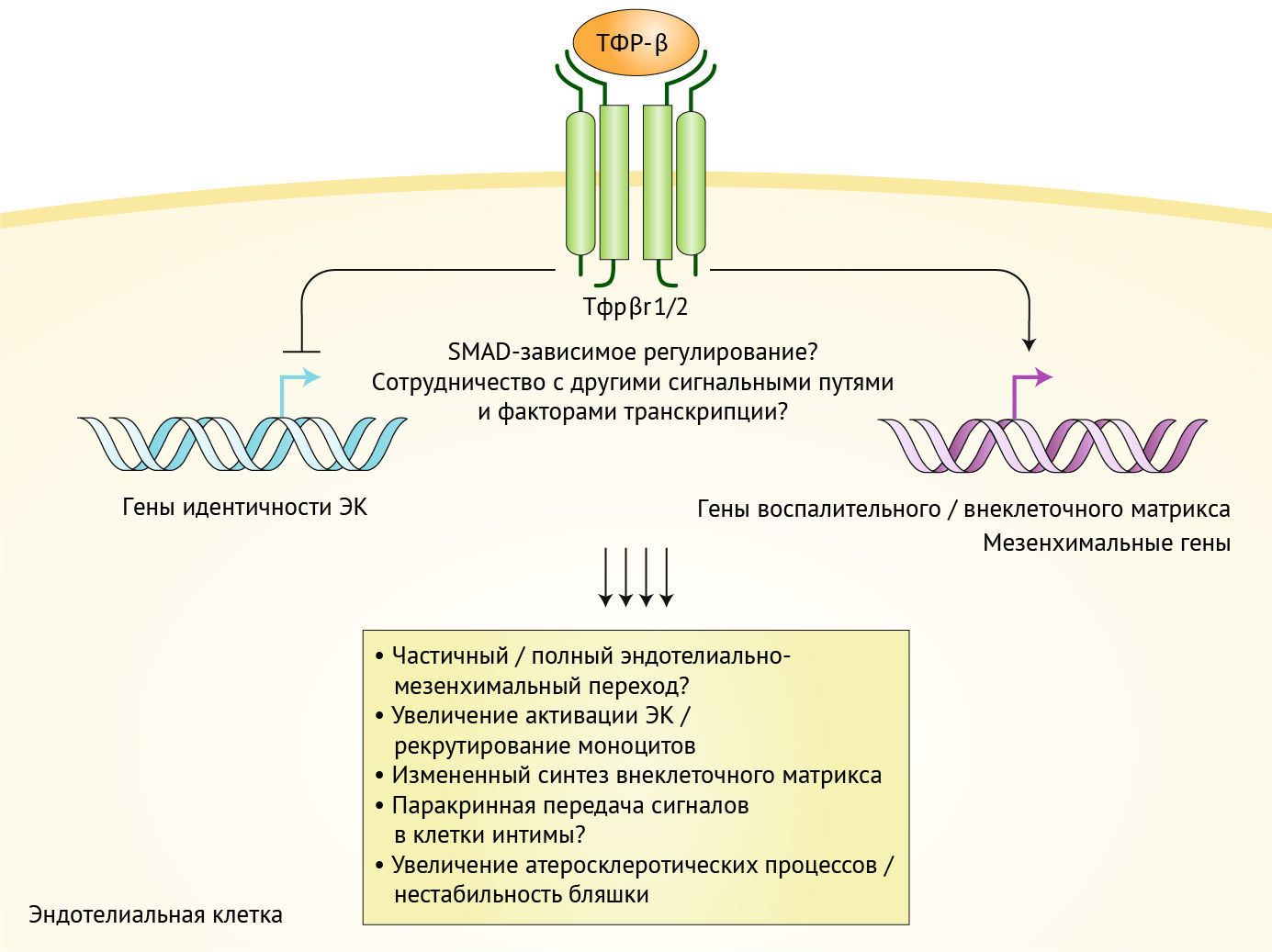
Рисунок 1
В выпуске Nature Metabolism Чен (Chen) и соавторы сообщают, что стимуляция ТФР-β однозначно индуцирует маркеры воспаления в культивируемых ЭК, но не в гладкомышечных клетках, Т-клетках или макрофагах. Специфически удаляя рецепторы ТФР-β 1 и 2 (Тфрβr1/2) в эндотелии перед воздействием на склонных к атеросклерозу мышей Apoe, выдерживая их на диете с высоким содержанием холестерина и жиров, авторы демонстрируют замедленное начало атеросклероза, уменьшение размера бляшек и количественного содержания в ней макрофагов, а также замедление образования некротического ядра.
Важность этого открытия была повышена также тем, что благодаря использованию модели установленного течения атеросклероза, авторы определили, что индуцибельная ЭК-специфическая делеция Тфрβr1/2 не только препятствует дальнейшему прогрессированию бляшек у мышей, получавших диету с высоким уровнем холестерина и содержанием жиров, но также усиливает регрессию бляшек, при переходе животных на нормальную диету. Кроме того, терапевтический подход, использующий ЭК-специфическую нанотерапию для доставки короткофокусной РНК с высоким сродством к Тфрβr1/2 на мышиной модели прогрессирования и регрессии атеросклероза, дал аналогичные обнадеживающие результаты. Таким образом, исследования показывают, что специфическая таргетная передача сигналов ТФР-β в эндотелии может способствовать регрессии бляшек.
Хотя воздействие на механизм атеросклероза носит отчасти провокационный характер, возможно, еще более интригующими являются результаты одноэлементного РНК-секвенирования аортальных «ЭК», полученных на этих моделях. Два кластера из всех клеток CD31+ характеризовались сниженным уровнем генов идентичности ЭК. Клетки обладали признаками трансформированного фенотипа EndMT, включая экспрессию мезенхимальных маркеров — генов, участвующих в модуляции внеклеточного матрикса и молекул с повышенной адгезией. В отсутствие передачи сигналов ТФР-β, эти EndMT и воспалительные признаки были снижены. Также идентифицированы дополнительные кластеры, представляющие частичные фенотипы EndMT, в которых клетки сохраняли маркеры ЭК, но коэкспрессировали мезенхимальные маркеры и имели провоспалительные генные сигнатуры. Опять же, устранение передачи сигналов ТФР-β, по-видимому, уменьшает эти признаки воспаления.
Таким образом, исследователи предоставили дополнительную информацию, доказывающую, что «трансформированный» эндотелиальный фенотип существует в атеросклеротических бляшках и может быть частично скорректирован, если передача сигналов ТФР-β будет блокироваться в эндотелии. Фундаментальные вопросы, касающиеся роли ТФР-β и EndMT в атеросклерозе еще предстоит рассмотреть: в какой степени расслоение и миграция клеток EndMT необходимы для влияния на процесс развития атеросклероза? Требуется ли полный EndMT или достаточно частичного EndMT? Является ли EndMT следствием или причиной провоспалительного фенотипа? В будущих исследованиях будет важно определить, влияет ли антагонизм передачи сигналов эндотелиального ТФР-β на миграцию эндотелиальных клеток в бляшку. По-прежнему считается, что экспрессия молекул адгезии, хемокинов или белков ЕСМ клетками, подвергающимися EndMT in situ, может модулировать нишу бляшек в отсутствии миграции путем усиления воспалительных клеток или изменения пролиферации и фенотипа клеток.
Наконец, стоит установить, может ли блокирование передачи сигналов ТФР-β в эндотелии быть эффективным при лечении других заболеваний, связанных с EndMT и воспалительным процессом, таких как фиброз внутренних органов, васкулопатия трансплантата или легочная артериальная гипертензия. Эта эндотелиальная сигнальная система ТФР-β вполне может быть критическим регуляторным механизмом, который можно использовать для лечения многих сосудистых воспалительных заболеваний.
Эндотелиальная передача сигналов ТФР-β управляет атерогенезом..
Передача сигналов через Тфрβr1/2 усиливает экспрессию воспалительных и мезенхимальных генов, модулируя при этом экспрессию генов ECM и подавляя гены идентичности ЭК. В результате эндотелиальные клетки активируются, проходят EndMT и способствуют образованию атерогенных поражений. Эта ТФР-β-опосредованная трансформация фенотипа ЭК, вероятно, влияет на поддержание моноцитов, а также на передачу сигналов в атеросклеротической бляшке. Было показано, что терапевтическое ингибирование передачи сигналов Тфрβr1/2 в ЭК, например, посредством доставки коротких интерферирующих РНК в эндотелий, вызывает регрессию атеросклеротических бляшек на мышиной модели.
