
Синдром Марфана

Синдром Марфана — наследственное заболевание соединительной ткани с аутосомно-доминантным типом наследования. В результате мутации нарушается синтез фибриллина, что ведет к аномальному строению α-цепи коллагена I типа, а также эластина, которые являются структурным компонентом сердечных клапанов, сердечной мышцы, стенок сосудов и опорно-двигательного аппарата [3].
На данный момент известно больше 500 мутаций гена фибриллина-1 (FBN1), но также заболевание может быть связано с деформациями генов FBN2, FBN3. Чаще всего возникают нарушения в процессе связывания кальция с фибриллином, в результате чего утрачивается устойчивость фибриллина к протеазам.
Около 15 лет назад был выделен синдром Марфана 2 типа, который обусловлен мутацией гена рецептора 2 трансформирующего ростового фактора β1. Клинически заболевание проявляется скелетными деформациями и расстройствами сердечно-сосудистой системы [3].
Клинические проявления синдрома Марфана могут быть как ярко выраженными, так и стертыми.
Люди, страдающие данным заболеванием, имеют высокий рост, арахнодактилию, скелетные деформации, патологии органа зрения, кардиоваскулярные патологии [4]. Часто новорожденные дети страдают недостаточностью клапанов сердца, дилатацией проксимальной части аорты. Нарушается формирование эластического каркаса аорты и аортальных клапанов с некрозом и фрагментацией эластических волокон, разрушаются коллагеновые волокна, происходит дистрофия гладкомышечных клеток с накоплением в них мукополисахаридов, разрастаются кисты, что приводит к острой сердечно-сосудистой недостаточности. В этом случае продолжительность жизни ребенка редко превышает один год [2].
Один из ведущих симптомов синдрома Марфана — поражение органа зрения (за исключением синдрома Марфана второго типа, где в клиническом статусе превалируют нарушения со стороны опорно-двигательного аппарата, сердечно-сосудистые расстройства, но отсутствуют поражения глаз) [1,3]. Патологоанатомически это определяется слабостью связочного аппарата хрусталика, что может привести к его вывиху. Также отмечаются слишком малые размеры и неправильная форма хрусталика, вследствие чего могут возникать деструкции и даже разрывы связок, что, в свою очередь, приводит к эктопии [1]. Еще одним симптомом является уменьшение диаметра сосудов, питающих сетчатку. Гетерохромия, мегалокорнеа, катаракта, гипоплазия радужной оболочки, цилиарной мышцы, пигментной каймы зрачков выявляются в 3 случаях из 10. Увеличение длины оси глазного яблока влечет за собой миопию и в дальнейшем может вызывать отслойку сетчатки [3].
У лиц, страдающих синдромом Марфана, относительно здоровых людей в популяции чаще выявляется недостаточная развитость ПЖК, поликистоз почек, паховые и бедренные грыжи, удвоение мочеточника и селезенки, деформации желчного пузыря. На рентгене органов грудной полости выявляются буллы и «капельное сердце», при этом восходящая аорта зачастую расширена [1].
На МРТ и КТ головы у более чем половины больных определяется диффузное утолщение твердой мозговой оболочки, дивертикулы паутинной, истончаются рога спинного мозга. При этом стоит отметить, что перечисленные выше заболевания прогрессируют с возрастом, что подтверждает динамическое наблюдение больных с синдромом Марфана [4].
Синдром Марфана классифицируют в зависимости от течения, генетических характеристик и клинических вариантов. Автором одной из классификации является Н. Т. Ватутинов [1].
.
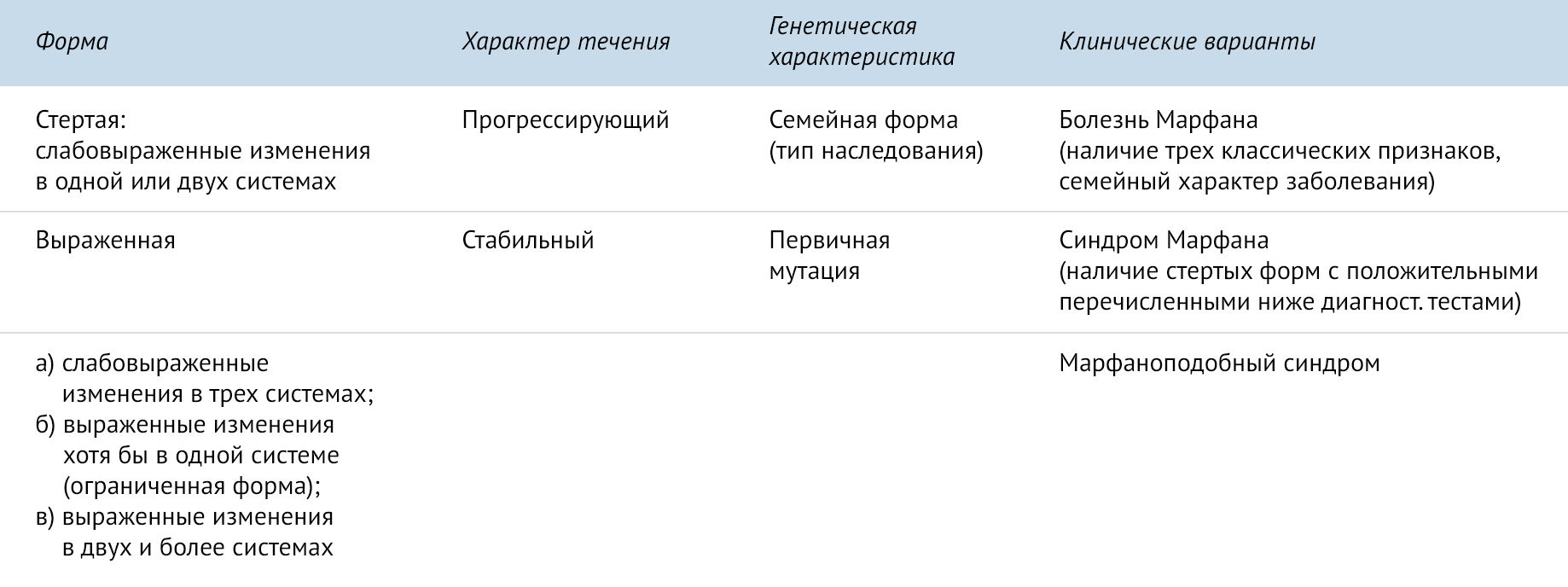
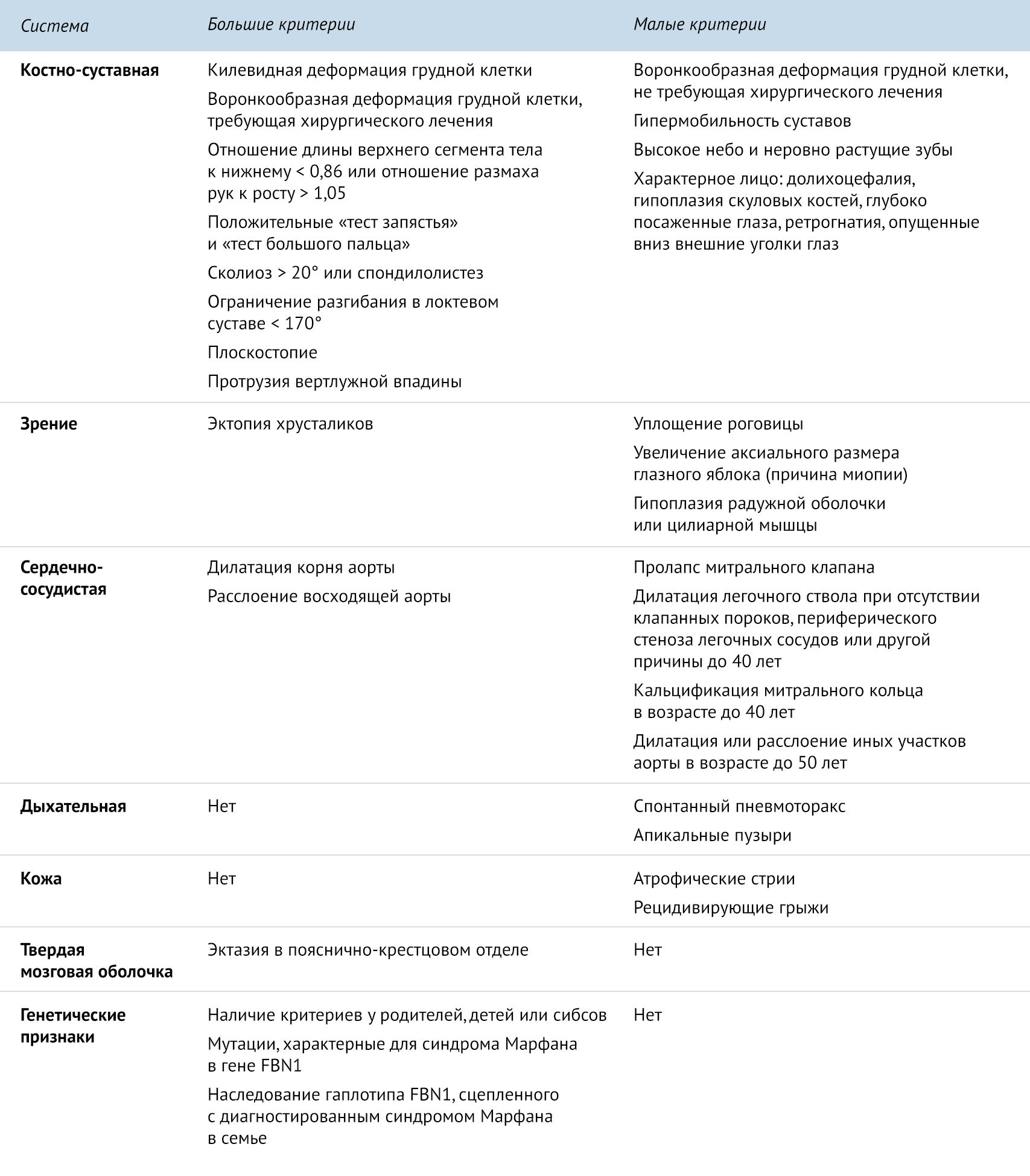
Данные критерии были пересмотрены, и в 1989 году были приняты так называемые Гентские критерии. Для постановки диагноза необходима совокупность двух больших критериев по двум системам и одного малого критерия по третьей системе [1].
.
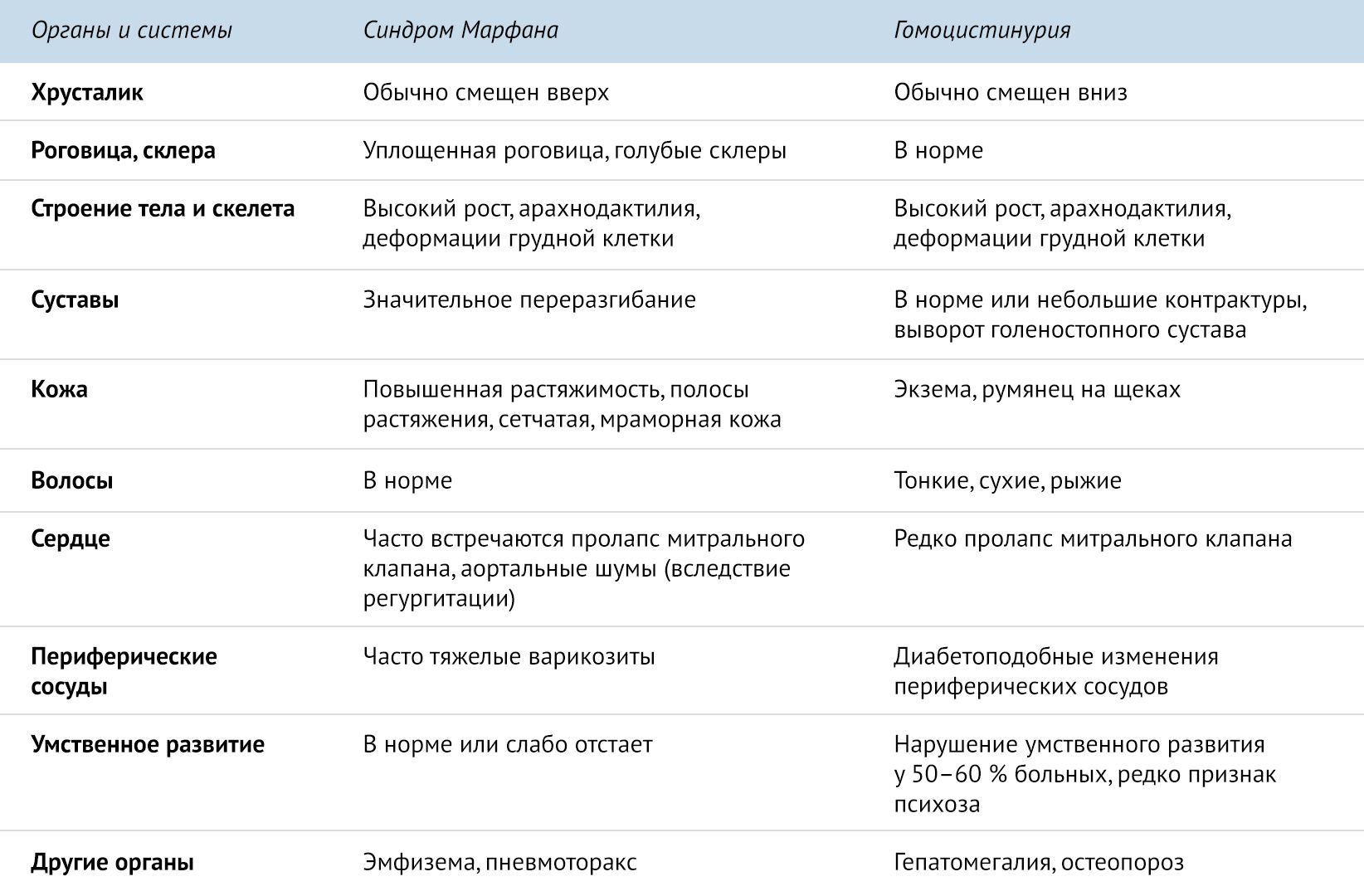
Синдром Марфана необходимо дифференцировать с аутосомно-рецессивным заболеванием — гомоцистинурией, типичным проявлением которого является повышенный уровень метионина в крови и гомоцистина в моче, что не встречается при синдроме Марфана [1].
.
Несмотря на то что синдром Марфана влечет за собой достаточно серьезные нарушения, пациенты, страдающие данным недугом, вполне способны поддерживать нормальное функционирование своих органов и систем в течении достаточно длительного времени. Для этого используются упражнения, направленные на укрепление мышц спины и брюшного пресса, а также связочного аппарата. Важным является и то, что занятия физкультурой позволяют поддерживать нормальный психический статус пациентов, а также стимулируют и укрепляют ССС. Показатели рекомендуемых нагрузок для таких пациентов составляют 25–50 Вт с увеличением ЧСС не более 110 уд/мин.
Лечение преимущественно направлено на общее укрепление организма [4].
Обязательным является сочетанием физических упражнений с общеукрепляющей медикаментозной терапией. Назначаются курсами по 2 месяца такие препараты, как терафлекс, диоксихолекальциферол, оксидевит, Кальций-Д3 Никомед. Обязательная терапия направлена на обогащение организма кальцием, магнием, фосфором, селеном. Лечение проводится с постоянным мониторированием уровня кальция и фосфора в крови и моче. Биохимический контроль показателей крови и мочи рекомендуется проводить не реже 1 раза в 2 недели. С целью улучшения усвоения аминокислот назначают анаболики [1].
Реабилитационная терапия проводится курсами не менее двух раз в год при контроле биохимических маркеров, таких как экскреция оксипролина (предшественник коллагена) и гликозаминогликанов с суточной мочой, уровень оксипролина и лизина в сыворотке крови, а также содержание кальция и фосфора в крови и моче, активность щелочной фосфатазы в крови. Диетотерапия проводится при наличии у больных поверхностного гастродуоденита, патологических рефлюксов, нарушения моторики на фоне повышения кислотообразующей функции желудка. Постоянный рацион питания должен быть богат продуктами, содержащими витамины А, Е, С, макро- и микроэлементы — кальций, медь, магний, железо, цинк, фосфор [1,4].
Источники:
- А.А. Тер-Галстян. Синдром Марфана./ А.А. Тер-Галстян, А.Р. Давтян. // Российский вестник перинатологии и педиатрии – 2008 – С. 58 – 64
- Daniel Judge. Marfan's syndrome. / Daniel Judge, Harry C. Dietz MD// Journal the Lancet – 2005 – C. 1034-1039
- Reed E. Pyeritz. The Marfan Syndrome: Diagnosis and Management . / Reed E. Pyeritz, M.D., Ph.D., and Victor A. McKusick, M.D. // The New England Journal of Medicine – 2015 – C. 85-96
- Reed E. Pyeritz. The Marfan’s syndrome. / Reed E. Pyeritz.//Annual Review of Medicine – 2009 – C. 31 - 34
