
Исследование происхождения SARS-CoV-2

В связи с эпидемией коронавируса, редакция Medical Channel приняла решения направить все свои силы на борьбу с этой угрозой. Мы переводим самые свежие и лучшие статьи из научных журналов, посвященные COVID-19, поскольку убеждены, что лучшим оружием являются знания. Если вы хотите поддержать нашу редакцию, чтобы выходило как можно больше переводов, то вы можете сделать пожертвование через Yandex или подписаться на наш Patreon. Будем очень признательны вашей помощи, все собранные средства будут идти на оплату работы переводчиков, редакторов и иллюстраторов.
Yandex: https://yasobe.ru/na/medach
Patreon: https://www.patreon.com/medach
Со времен появления первых сообщений о новом виде пневмонии (COVID-19) в городе Ухань, провинции Хубэй, Китай, ведутся серьезные дискуссии о теории происхождении возбудителя — вируса SARS-CoV-2 (также называемого HCoV-19). Инфекции, вызванные SARS-CoV-2, в настоящее время широко распространены, и по состоянию на 11 марта 2020 года было подтверждено 121 564 случая в более чем 110 странах, при этом 4373 случая заболевания оказались смертельными.
SARS-CoV-2 является седьмым коронавирусом, который достоверно вызывает какие-либо заболевания у человека. SARS-CoV, MERSCoV и SARS-CoV-2 ответственны за тяжелые респираторные проявления заболевания, тогда как HKU1, NL63, OC43 и 229E связаны с легкими симптомами. Здесь мы рассмотрим известные данные исследований, которые позволят сделать выводы о происхождении SARS-CoV-2 из сравнительного анализа геномных данных. Мы предлагаем обзор отличительных особенностей генома SARS-CoV-2 и обсуждаем сценарии, при которых данные особенности могли возникнуть. Наши анализы ясно показывают, что SARS-CoV-2 не является искусственной лабораторной конструкцией или целенаправленно управляемым вирусом.
Примечательные особенности генома SARS-CoV-2
Наше сравнение альфа- и бета-коронавирусов выявляет две заметные геномные особенности SARS-CoV-2:
- на основе структурных исследований и биохимических экспериментов SARS-CoV-2, по-видимому, более оптимизирован для связывания с человеческим рецептором ACE2, нежели другие вирусы этого семейства;
- белок-шип SARSCoV-2 имеет функциональный сайт многоосновного (фуринового) расщепления на границе S1-S2 посредством вставки 12 нуклеотидов, что дополнительно привело к приобретению трех O-связанных гликанов вокруг сайта.
Мутации в рецептор-связывающем домене SARS-CoV-2
Рецептор-связывающий домен (RBD) в белке шипа является наиболее вариабельной частью генома коронавируса. Было показано, что шесть аминокислот RBD являются критическими для связывания с рецепторами ACE2 и для определения диапазона хозяев для различных SARS-CoV-подобных вирусов с координатами, основанными на SARS-CoV, — это Y442, L472, N479, D480, T487 и Y4911, которые соответствуют L455, F486, Q493, S494, N501 и Y505 в SARS-CoV-27. Пять из этих шести остатков различаются между SARSCoV-2 и SARS-CoV (рис. 1а). На основании структурных исследований и биохимических экспериментов SARS-CoV-2, по-видимому, имеет RBD, который обладает высоким сродством к ACE2 у человека, хорьков, кошек и других видов с высокой гомологией рецепторов.
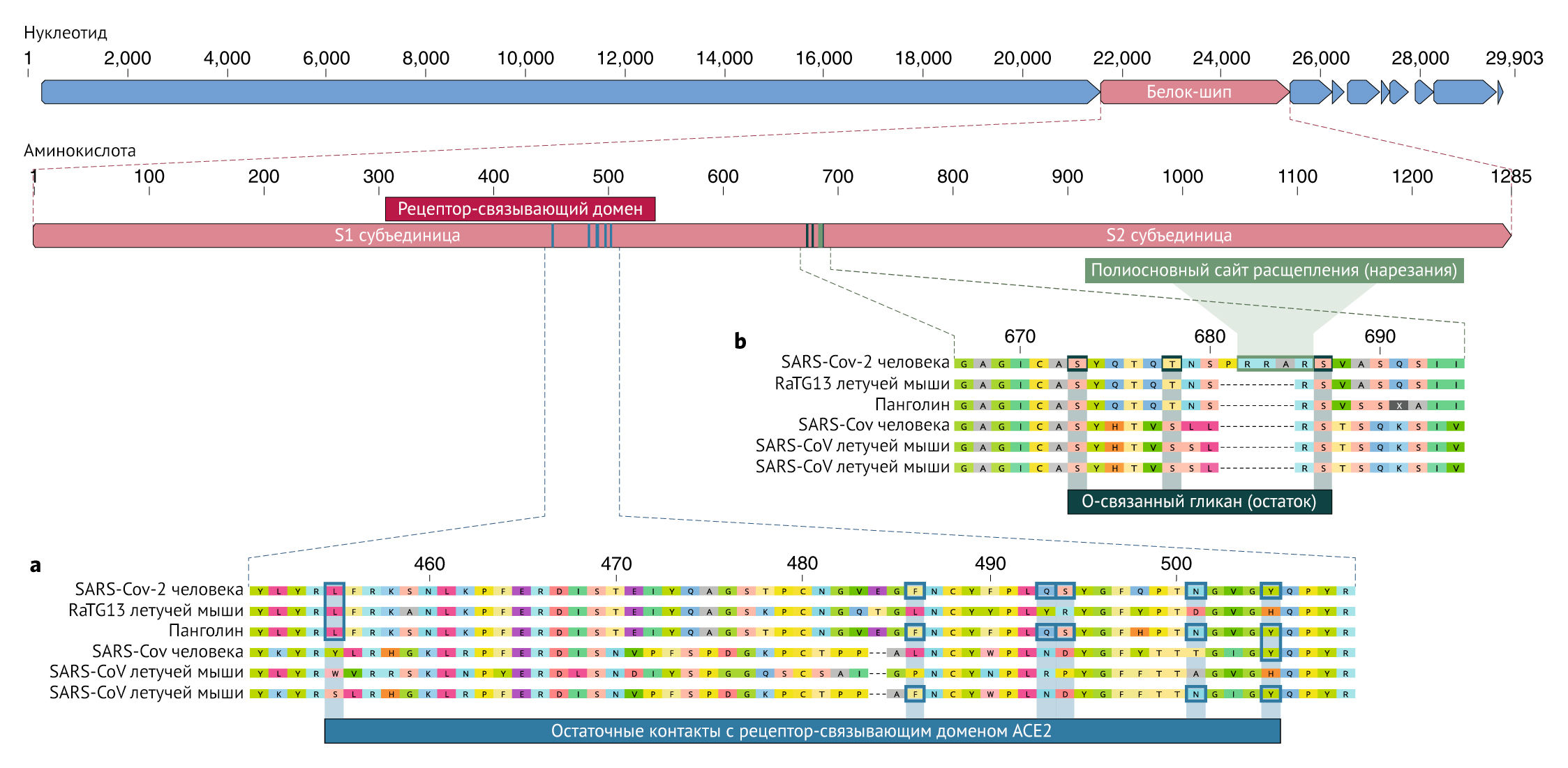
b) Приобретение многоосновного сайта расщепления и О-связанных гликанов. Как сайт многоосновного расщепления, так и три соседних О-связанных гликана являются уникальными для SARS-CoV-2 и ранее не наблюдались в бета-коронавирусах линии В. Показаны последовательности из NCBI GenBank, коды доступа MN908947, MN996532, AY278741, KY417146 и MK211376. Последовательности коронавируса панголина представляют собой некий микс, полученный из SRR10168377 и SRR10168378 (NCBI BioProject PRJNA573298).
Хотя приведенный выше анализ предполагает, что SARS-CoV-2 может связывать человеческий ACE2 с более высокой аффинностью, вычислительный анализ предсказывает, что его взаимодействие с рецепторами сложно назвать идеальным, и что последовательность RBD отличается от показанной в SARS-CoV, чтобы быть более «удобной» для связывания с рецептором. Таким образом, высокоаффинное связывание белка шипа SARS-CoV-2 с человеческим ACE2, скорее всего, является результатом естественного отбора на человеческом или подобном человеку рецептору ACE2. Это убедительное доказательство того, что SARS-CoV-2 не является продуктом целенаправленных манипуляций.
Сайт расщепления многоосновного фурина и О-связанные гликаны
Второй примечательной особенностью SARS-CoV-2 является многоосновный сайт расщепления (RRAR) на стыке S1 и S2, двух субъединиц шипа (Рис. 1b). Он позволяет эффективно расщепляться фурином и другими протеазами и играет роль в повышении вирулентности и наличие широкого диапазона выбора организмов-хозяев. Кроме того, ведущий пролин также вставлен в этот сайт в SARS-CoV-2. Таким образом, вставленная последовательность является PRRA (Рис. 1b). Предполагается, что поворот, созданный пролином, приведет к добавлению O-связанных гликанов к S673, T678 и S686, которые фланкируют сайт расщепления и являются уникальными именно для SARS-CoV-2 (Рис. 1b). Подобные сайты многоосновного расщепления не наблюдались ни в каких родственных «бета-коронавирусах линии В», хотя другие бета-коронавирусы человека, включая HKU1 (линия А), имеют эти сайты и предсказывают О-связанные гликаны. Учитывая уровень генетической изменчивости в шипе, вероятно, SARS-CoV-2-подобные вирусы с частичными или полными многоосновными сайтами расщепления будут позже обнаружены и у других видов. Функциональные последствия многоосновного сайта расщепления в SARS CoV-2 неизвестны, и будет важно определить его влияние на трансмиссивность и патогенез заболевания на животных моделях.
Эксперименты с SARS-CoV показали, что вставка сайта расщепления фурином в месте соединения S1-S2 усиливает клеточное слияние, не влияя на уровень проникновения вируса. Кроме того, эффективное расщепление шипа MERS-CoV позволяет MERS, подобным коронавирусам от летучих мышей, инфицировать и клетки человека. В случае вирусов птичьего гриппа быстрая репликация и передача в очень плотных популяциях кур стала результатом приобретения многоосновных сайтов расщепления в белке гемагглютинином (HA), который выполняет функцию, аналогичную функции белка шипа коронавируса. Приобретение многоосновных сайтов расщепления в HA путем вставки или рекомбинации превращает вирусы птичьего гриппа с низкой патогенностью в высокопатогенные формы. Получение многоосновных сайтов расщепления HA также наблюдалось после повторного пассажа в клеточной культуре или через животных. Функция О-связанных гликанов в новом коронавирусе пока неясна, но они могут создавать «муциноподобный домен», который экранирует эпитопы или ключевые остатки белка-шипа SARS-CoV-2. Некоторые вирусы используют муциноподобные домены в качестве щитов гликана, связанных с ускользанием от иммунного ответа. Хотя наличие О-связанного гликозилирования в коронавирусах является лабораторно достоверным фактом, необходимы экспериментальные исследования, чтобы определить, используются ли эти сайты в SARS-CoV-2.
Теории происхождения SARS-CoV-2
Маловероятно, что SARS-CoV-2 появился в результате лабораторных манипуляций с неким родственным SARS-CoV-подобным коронавирусом. Как отмечено выше, RBD SARS-CoV-2 оптимизирован для связывания с человеческим ACE2 с помощью естественного отбора, то есть механизма, отличного от ранее предсказанного. Кроме того, если бы была проведена генетическая манипуляция, вероятно, была бы использована одна из нескольких обратных генетических систем, доступных для бета-коронавирусов. Тем не менее, генетические данные неопровержимо показывают, что SARSCoV-2 не получен из каких-либо ранее использованных вирусных магистралей. Вместо этого мы предлагаем два сценария, которые могут правдоподобно объяснить происхождение SARS-CoV-2:
- мутации в результате естественного отбора у животного-хозяина, предшествующие зоонозному переносу;
- естественный отбор у людей уже после зоонозного переноса;
- мы также обсуждаем, могли ли мутации в результате естественного отбора во время переноса вызвать SARS-CoV-2.
Сценарий 1:
Естественный отбор у животного-хозяина перед зоонозным переносом
Так как многие ранние случаи COVID-19 были связаны с рынком Хуанань города Ухань, Китай, возможно, что источник инфекции в виде животных присутствовал именно в этом месте. Учитывая сходство SARSCoV-2 с SARS-CoV-подобными коронавирусами летучих мышей, вполне вероятно, что летучие мыши служат резервуарными хозяевами для своего предшественника. Хотя RaTG13, отобранный у летучей мыши Rhinolophus affinis, примерно на 96% идентичен SARS-CoV-2, его шип расходится в RBD, что позволяет предположить, что он не может эффективно связываться с ACE27 человека (Pис. 1a).
Малайские панголины (Manis javanica), незаконно ввезенные в провинцию Гуандун, переносили коронавирусы, похожие на SARSCoV-2. Хотя вирус летучей мыши RaTG13 остается наиболее близким к SARS-CoV-2 по характеру своего генома, некоторые коронавирусы панголина демонстрируют сильное сходство с SARS-CoV-2 в RBD, включая все шесть ключевых сайтов RBD (рис. 1). Это ясно показывает, что связывающий белок SARS-CoV-2, оптимизированный для связывания с человеческим ACE2, является результатом естественного отбора. Ни у бета-коронавирусов летучих мышей, ни у бета-коронавирусов панголина, выделенных и изученных до настоящего времени, нет многоосновных сайтов расщепления. Хотя не было выявлено ни одного коронавируса животных, который был бы достаточно похожим, чтобы быть прямым предшественником SARS-CoV-2. Разнообразие коронавирусов у летучих мышей и других видов подвергается многочисленным исследованиям. Как инсерции, так и делеции могут происходить вблизи S1-S2 соединения коронавирусов, что показывает, что сайт многоосновного расщепления может возникать в результате естественного эволюционного процесса. Для того, чтобы вирус-предшественник приобрел как сайт многоосновного расщепления, так и мутации в связывающем белке, подходящем для связывания с человеческим АСЕ2, животному-хозяину, вероятно, потребуется высокая плотность популяции (чтобы обеспечить эффективный естественный отбор для проведения) и кодирование АСЕ2 гена, похожего на человеческий ортолог.
Сценарий 2:
Естественный отбор, происходящий в организме человека после зоонозного переноса
Вполне возможно, что предок SARS-CoV-2 попал в организм человека, приобретя геномные характеристики, описанные выше, путем адаптации во время передачи от человека к человеку. После того, как эти изменения были получены, они позволили пандемии развиться и создали достаточно большое количество прецедентов, чтобы запустить систему наблюдения, которая и обнаружила новый вирус. Все секвенированные до настоящего времени геномы SARS-CoV-2 имеют генетические признаки, описанные выше, и, таким образом, происходят от общего предка, который также обладал данными признаками. Присутствие в панголинах RBD, очень похожего на SARS-CoV-2, позволяет сделать вывод, что это также было возможно в вирусе, который стал распространяться и на людей. Оценки времени происхождения последнего общего предка SARS-CoV-2, сделанные с использованием текущих данных о вирусе, указывают на появление возбудителя в конце ноября 2019 года – начале декабря 2019 года, что согласуется с самыми ранними ретроспективно подтвержденными случаями. Следовательно, этот сценарий предполагает период нераспознанной передачи у человека между начальным зоонозным событием и приобретением многоосновного сайта расщепления. По сути, эта ситуация характерна для MERS-CoV, для которой все случаи заболевания людей являются результатом повторного распространения вируса с верблюдов-дромадеров, приводящих к спорадическим инфекциям или коротким цепям передачи, которые в конечном итоге проходят без адаптации к дальнейшей устойчивой передаче. Исследования полученных от человека образцов вируса могут дать информацию о том, как именно произошло его распространение и эволюция. Ретроспективные серологические исследования также могут быть информативными, поэтому было проведено несколько подобных исследований, показывающих низкую активность SARSCoV-подобных коронавирусов в некоторых районах Китая. Однако эти исследования не смогли критически ответить на вопрос, были ли воздействия вызваны предшествующими инфекциями SARS-CoV, SARS-CoV-2 или другими SARSCoV-подобными коронавирусами. Дальнейшие серологические исследования должны проводиться для определения степени предшествующего воздействия SARS-CoV-2 на человека.
Сценарий 3:
Мутации в вирусе возникли в результате естественного отбора во время переноса
Фундаментальные исследования, связанные с пассажем SARS-CoV-подобных коронавирусов летучих мышей в клеточных культурах и/или на животных моделях, ведутся в течение многих лет в лабораториях уровня 2 по биобезопасности во всем мире, и имеются документально подтвержденные случаи лабораторных утечек SARS-CoV. Поэтому мы должны изучить возможность непреднамеренного лабораторного высвобождения SARS-CoV-2. Теоретически, возможно, что SARS-CoV-2 приобрел мутации RBD (Рис. 1a) во время адаптации к пассажу в культуре клеток, как это наблюдалось ранее в исследованиях SARS-CoV. Однако обнаружение SARS CoV-подобных коронавирусов у панголинов с почти идентичными RBD обеспечивает гораздо более логичное объяснение того, как SARS-CoV-2 приобретал их посредством рекомбинации или мутации. Приобретение как многоосновного сайта расщепления, так и O-связанных гликанов также противоречит сценариям, основанным на исследованиях в культуре клеток. Новые многоосновные сайты расщепления наблюдались только после длительного прохождения вируса птичьего гриппа с низкой патогенностью in vitro или in vivo. Кроме того, гипотетическая генерация SARS-CoV-2 путем клеточной культуры или пассажа у животных потребовала бы предварительного выделения вируса-предшественника с очень высоким генетическим сходством, которое пока не было описано. Последующее образование многоосновного сайта расщепления потребовало бы повторного пассажа в клеточной культуре или у животных с рецепторами ACE2, сходными с таковыми у людей, но такое исследование также ранее не было проведено. Наконец, генерация О-связанных гликанов также вряд ли произошла из-за пассажа в клеточной культуре, поскольку такие особенности предполагают участие иммунной системы, что невозможно в условиях клеточной культуры.
Заключение и выводы
В разгар глобальной чрезвычайной ситуации в области общественного здравоохранения разумно задаться вопросом о причинах возникновения пандемии. Детальное понимание того, как вирус животных широким шагом перешел через границы видов для столь активного заражения людей, поможет предотвратить будущие случаи зоонозного переноса. Например, если SARS-CoV-2 предварительно адаптирован к другим видам животных, то существует риск его повторного появления. Напротив, если адаптивный процесс произошел у людей, то даже если происходят повторные зоонозные переносы, они вряд ли начнут развиваться без той же серии мутаций. Кроме того, выявление ближайших родственников вируса SARS-CoV-2, циркулирующего у животных, будет в значительной степени способствовать изучению морфологии вируса, вирулентности и разработки тактики эффективной терапии. Действительно, наличие последовательности RaTG13 у летучих мышей помогло выявить ключевые мутации RBD и сайт многоосновного расщепления.
Особенности генома нового коронавируса, описанные в данном исследовании, могут частично объяснить вирулентность и контагиозность SARS-CoV-2 у людей. Хотя данные показывают, что SARSCoV-2 не является целенаправленно управляемым вирусом, в настоящее время невозможно доказать или опровергнуть другие теории его происхождения, описанные здесь. Однако поскольку мы наблюдали все заметные признаки SARS-CoV-2, включая оптимизированный RBD и многоосновный сайт расщепления, в связанных коронавирусах в природе, мы не считаем, что какой-либо искусственный лабораторный сценарий правдоподобен. Большее количество исследований может изменить баланс данных в пользу одной гипотезы по отношению к другой. Получение родственных вирусных последовательностей из животных источников было бы наиболее точным способом выявления вирусного происхождения. Например, будущее наблюдение за промежуточным или полностью сформированным многоосновным сайтом расщепления у вируса, подобного SARS-CoV-2 у животных, могло бы еще больше подкрепить гипотезы естественного отбора. Было бы также полезно получить больше генетических и морфофункциональных данных о SARSCoV-2, включая исследования на животных моделях. Идентификация потенциального промежуточного хозяина SARS-CoV-2, а также определение последовательности вируса в очень ранних случаях были бы весьма информативными. Независимо от точных механизмов, с помощью которых SARSCoV-2 возник в результате естественного отбора, постоянное наблюдение за пневмонией у людей и других животных, несомненно, имеет огромное значение.
