
Вступая в современную эру генной терапии

Аннотация
Генная терапия набирает обороты: растет число успешных клинических исследований, и всё больше препаратов получают одобрение для лицензирования. Исследователи (с определенной степенью осторожности) надеются, что эффективные, надежные и безопасные методы лечения принесут пользу пациентам не только с моногенными нарушениями, но и с комплексными приобретенными заболеваниями. На данный момент ряд новых подходов уже находится на этапе внедрения в клинику, однако этот обзор посвящен клиническим исследованиям, создавшим область генной терапии в её текущем состоянии.
Введение
Генная терапия представляет собой введение больному генетического материала с целью лечения заболевания. Хотя такая концепция существовала на протяжении многих десятилетий, клинические исследования начались с 1990 года, когда в Национальном институте здравоохранения США было проведено первое клиническое исследование терапии редкого иммунодефицитного состояния. С тех пор было начато более 2500 клинических исследований широкого спектра направленности: от различных моногенных заболеваний до инфекций, комплексных нейродегенеративных заболеваний и рака (рис. 1).
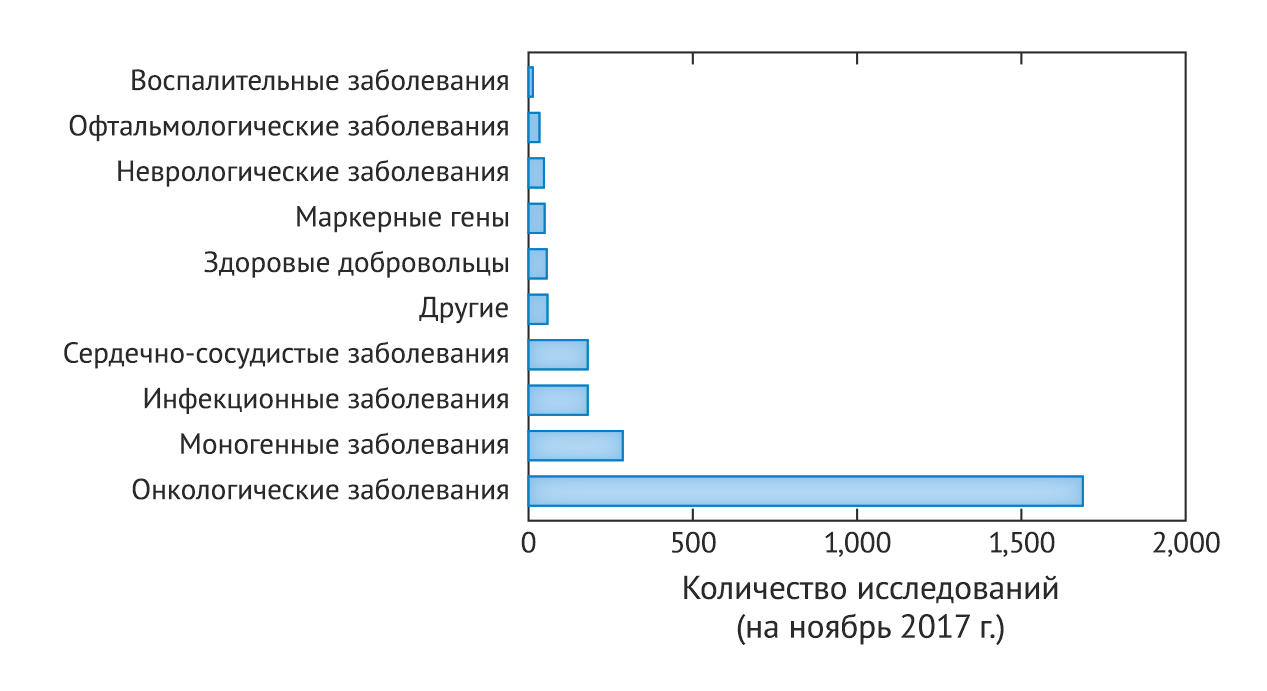
Адаптировано из источника 1.
Генную терапию можно классифицировать несколькими способами, например, по типу заболевания (генетическое нарушение или комплексное приобретенное состояние), по характеристикам векторной системы, т. е. по средству доставки гена (интегративный или неинтегративный), а также по тому, каким образом осуществляется введение вектора — in vivo (непосредственно пациенту) или ex vivo (во взятые у пациента и культивированные клетки, которые впоследствии пересаживают обратно).
В своем самом прямом воплощении цель генной терапии генетических заболеваний заключается в долговременном поддержании экспрессии введенного гена на уровне, достаточно высоком для достижения терапевтического эффекта (т. н. усилительная генная терапия, augmentation gene therapy). Чаще всего вводимый ген представляет собой нормальную копию мутировавшего у пациента гена. Также можно подавить экспрессию вредоносного гена, применив технологии РНК-интерференции или инструменты редактирования генома. Теоретически, терапия с применением вышеописанных технологий возможна, однако пока клинические испытания с исправлением мутировавшего гена в месте его точного расположения посредством гомологичной рекомбинации с донорным шаблоном (или путём редактирования оснований) не проводились [2] (рис. 2).
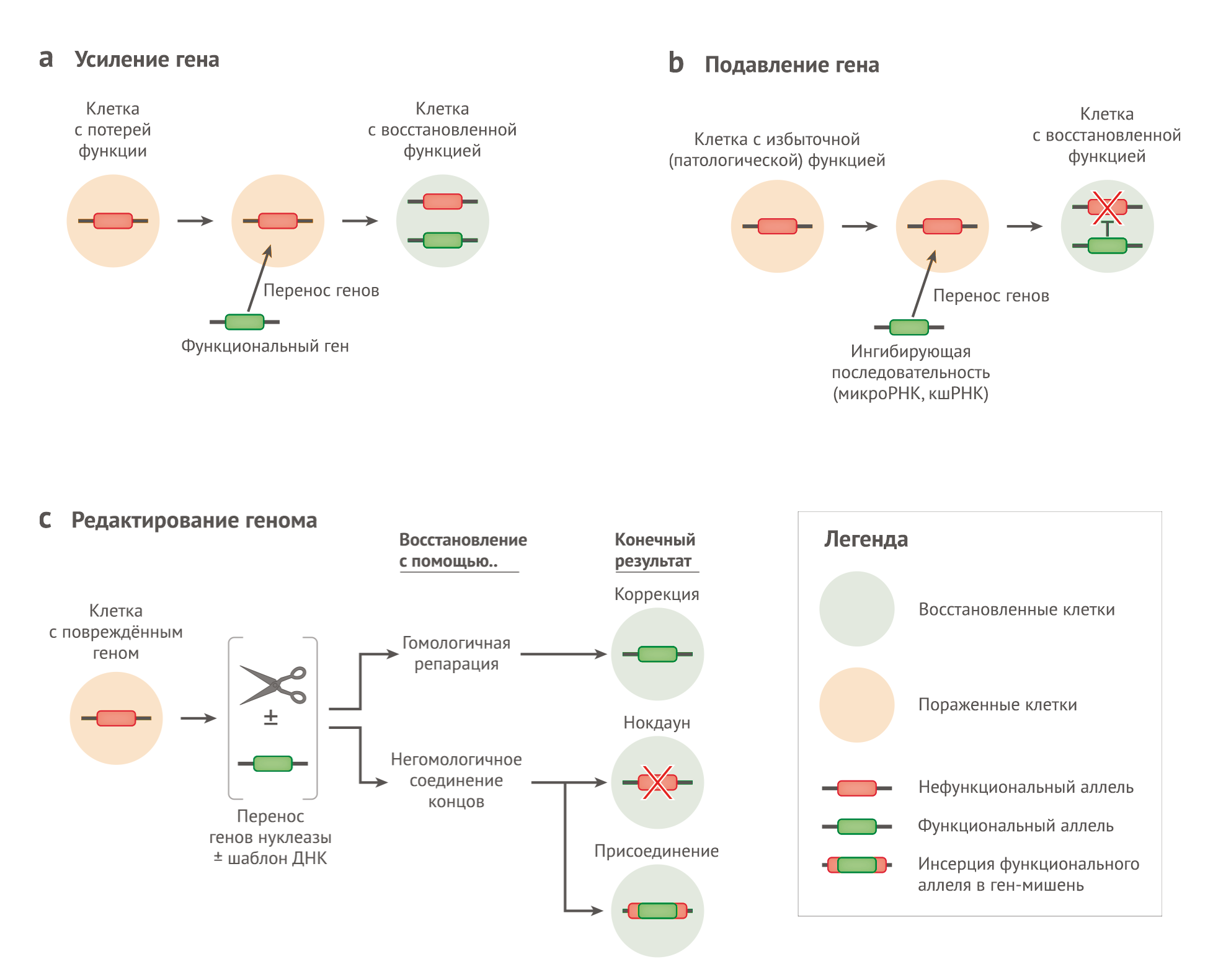
(b) При некоторых других заболеваниях, таких как болезнь Гентингтона, определенные функции клеток утрачиваются из-за накопления токсичного дефектного белка. Подавление гена направлено на восстановление функционального состояния клеток путем уменьшения экспрессии мутировавшего гена с помощью РНК-интерференции.
(c) Хотя редактирование генома не обязательно требует использования нуклеазы, эффективность ген-специфического редактирования в клетках млекопитающих обычно повышается за счет индукции двухцепочечного разрыва ДНК в сайте-мишени. Выбор конкретного механизма репарации ДНК будет определять исход редактирования генома. В своей простейшей форме, образовавшийся разрыв ДНК устраняется путём негомологичного соединения концов, которое может привести к нокдауну гена в том случае, если репарация прошла неидеально. В присутствии экзогенного шаблона, кодирующего функциональный ген, репарация ДНК также может привести к коррекции мутировавшего гена in situ посредством гомологичной рекомбинации. Третьим потенциальным результатом является вставка шаблона ДНК посредством негомологичного соединения концов, что приведет к присоединению нормального гена, а не к его коррекции.
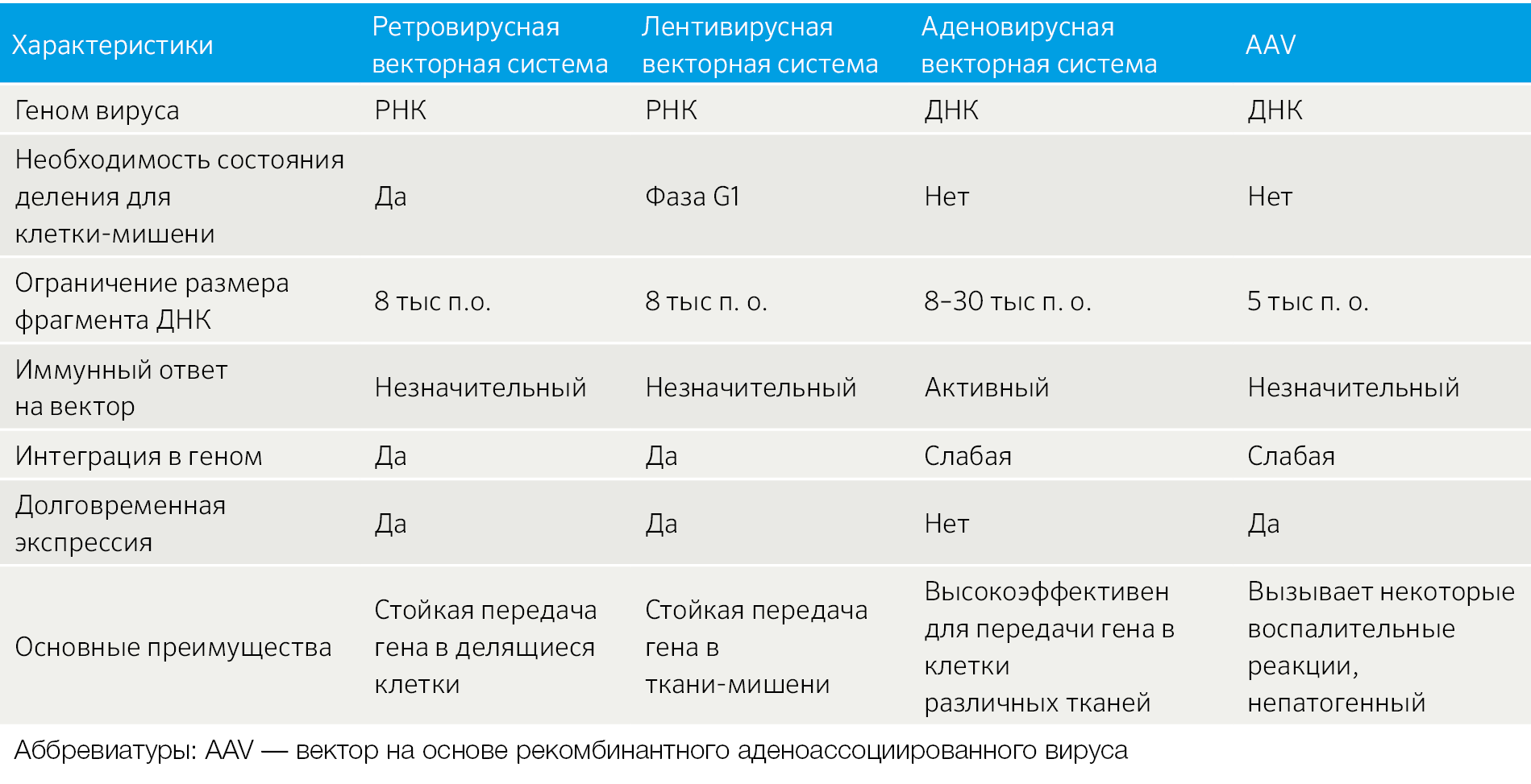
Одним из основных факторов, определяющих различия между терапией in vivo и ex vivo, является выбор векторной системы (Табл. 1). Как отмечено выше, векторы можно разделить на преимущественно интегрирующие и преимущественно неинтегрирующие. При введении генетического материала в стволовые клетки очень важно использовать интегрирующие векторы, поскольку в таком случае донорская ДНК внедрится в геном стволовой клетки, будет реплицироваться при делении и, таким образом, передается во все дочерние клетки. Генная терапия in vivo часто нацелена на долгоживущие постмитотические клетки. В этих клетках, поскольку они больше не делятся, экспрессия гена может поддерживаться в течение долгого времени, пока стабильна введённая ДНК (для поддержания экспрессии гена на протяжении жизни клетки применяется стабилизация вектора в виде эписом — суперскрученных кольцевых молекул ДНК). Большинство стратегий генной терапии для лечения генетических заболеваний в настоящее время используют два типа векторных систем: лентивирусные векторы для переноса генов ex vivo в гемопоэтические и другие стволовые клетки [3] и векторы на основе рекомбинантного аденоассоциированного вируса (AAV) для переноса генов in vivo в постмитотические типы клеток [4].
Таблица 1 | Вирусные векторные системы, рассмотренные в данном обзоре
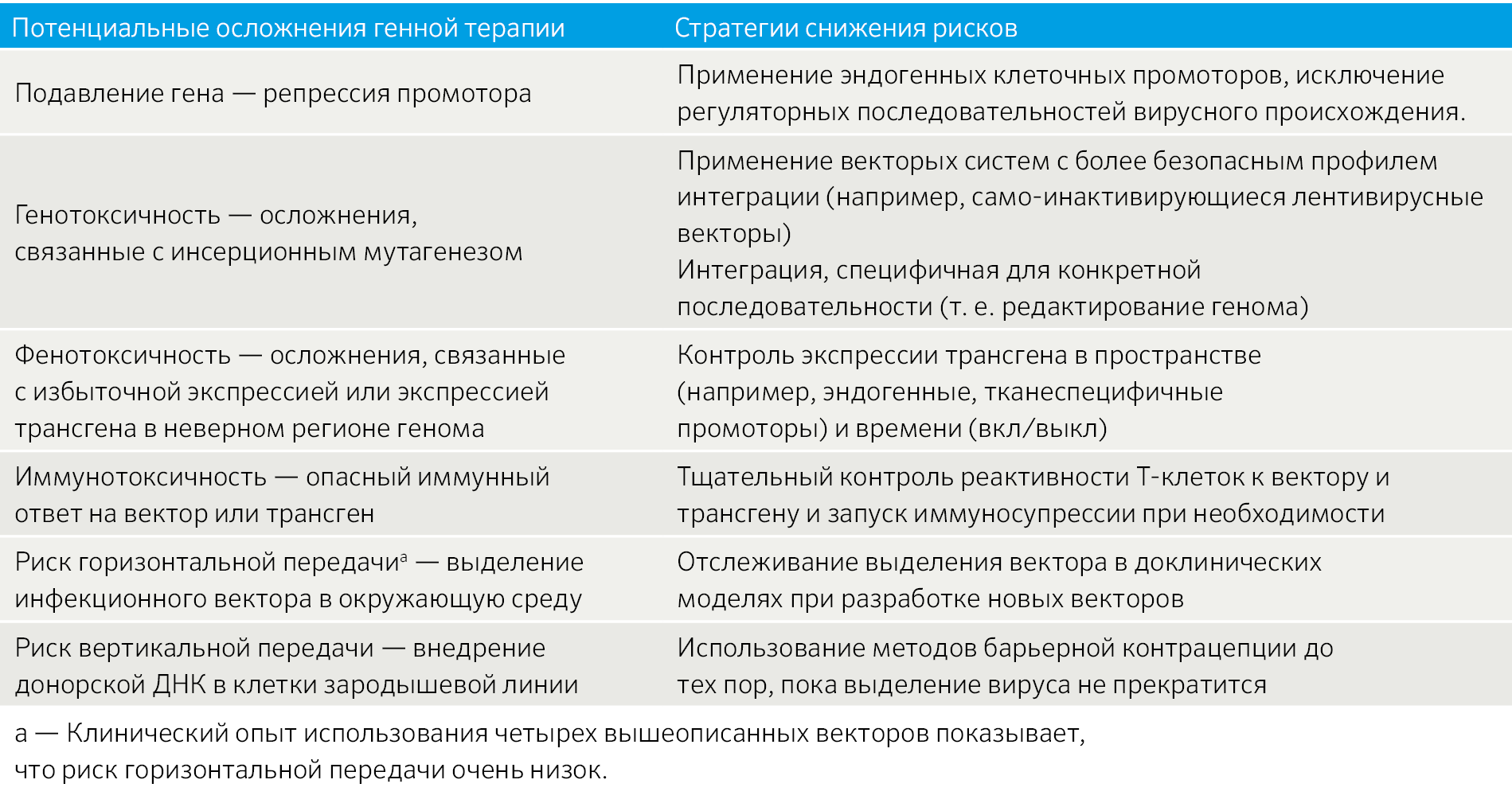
Потенциальные риски генной терапии для лечения генетических заболеваний рассматривались исследователями ещё до начала её применения в клинике (табл. 2). По мере накопления данных с 1990-х годов стало очевидно, что риски делятся на две основные группы. Применение первых интегрирующих векторов было сопряжено с риском инсерционного мутагенеза, что подтверждалось развитием Т-клеточного лейкоза у детей, проходивших лечение от Х-сцепленного тяжелого комбинированного иммунодефицита (SCID) [5]; а в случае введения вектора in vivo возникал риск неблагоприятных иммунных реакций. Пока применение одних векторов оказалось опасным для жизни пациентов [6], для других под угрозой находилась их долговременная эффективность, но не безопасность [7]. Так, генная терапия рака часто связана с риском чрезмерной активации Т-лимфоцитов.
Первоначальный успех генной терапии был связан со стратегией ex vivo, в которой больным с первичными иммунодефицитами трансплантировали их собственные (аутологичные) гемопоэтические стволовые клетки (ГСК), несущие скорректированный ген. Генная терапия in vivo отставала, но казалось, что успех непременно будет достигнут к 2000 году. Однако, несколько нашумевших побочных явлений терапии, включая злокачественную трансформацию клеток из-за инсерционного мутагенеза при действии интегрирующих векторов и смерть пациента в клиническом испытании аденовирусного вектора повлияли как на готовность врачей и пациентов участвовать в испытаниях, так и на готовность инвесторов поддержать дальнейшую работу в этой области. Возрождение генной терапии примерно через десять лет во многом было связано с успехами клинических испытаний, в которых использовались стратегии как ex vivo (для Х-сцепленного SCID, дефицита аденозиндезаминазы и адренолейкодистрофии), так и подходы in vivo (врожденный амавроз Лебера типа 2 и гемофилия B).
Таблица 2 | Потенциальные осложнения генной терапии, применяемой в клинике
На сегодняшний день в странах Запада одобрение получили шесть препаратов для генной терапии. Glybera®, терапия на основе AAV для лечения семейного дефицита липопротеин-липазы, в 2012 году получила условное маркетинговое одобрение Европейского агентства по лекарственным средствам (European Medicines Agency, EMA). IMLYGIC®, генетически модифицированный вирус простого герпеса типа 1 для местного лечения неоперабельных поражений у пациентов с меланомой, был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в 2015 году. Strimvelis®, терапия на основе γ-ретровируса для лечения тяжелого комбинированного иммунодефицита, вызванного дефицитом аденозиндезаминазы (ADA-SCID), получила одобрение EMA в 2016 году. Три препарата для генной терапии были одобрены FDA в 2017 году. KYMRIAH® и YESCARTA® представляют собой препараты для CAR-T-клеточной терапии (Chimeric Antigen Receptor T-Cell или T-клетки с химерным антигенным рецептором), а именно генетической модификации собственных CD19-клеток пациента. Оба препарата показаны для лечения неходжкинской лимфомы, а KYMRIAH® — и для лечения острого лимфобластного лейкоза. LUXTURNA® представляет собой генную терапию на основе AAV и предназначена для лечения дистрофии сетчатки, связанной с биаллельной мутацией RPE65.
Целью данного обзора является создание основы для оценки клинических результатов текущих исследований, поскольку число отчетов и статей продолжает увеличиваться. Несколько новых препаратов в настоящее время находятся на стадии рассмотрения заявки на регистрацию, и в связи с этим представляется вероятным, что у врачей появится всё больше возможностей для собственной оценки генной терапии как метода лечения.
Генная терапия ex vivo
Генная терапия в гемопоэтических стволовых клетках
Подходы к генной терапии ex vivo для лечения генетических заболеваний в основном нацелены на ГСК — самообновляющиеся клетки, дающие начало всем клеточным росткам крови. Эти клетки активно делятся с целью самообновления своей популяции или же для дифференцировки в определенный тип клеток, поэтому для того, чтобы терапевтический ген присутствовал во всех дочерних клетках, он должен быть стабильно интегрирован в геном.
В конце 1990-х годов клиническое исследование Fischer с коллегами [8], впервые убедительно продемонстрировало терапевтический эффект переноса гена для лечения Х-сцепленного SCID (SCIDX1). Это аутосомно-рецессивное заболевание возникает из-за мутаций в гене, кодирующем γc-субъединицу цитокинового рецептора, что приводит к отсутствию циркулирующих Т-клеток и естественных киллеров (NK), а также нарушению функций В-лимфоцитов. От гибели в первые годы жизни больного может спасти только успешная трансплантация костного мозга, которая восстанавливает нормальную работу иммунной системы. Оригинальные клинические исследования генной терапии SCIDX1 при помощи γ-ретровирусных векторов первого поколения, в которых ген экспрессировался под контролем вирусных регуляторных элементов, показали многообещающие признаки эффективности такого метода, а именно успешную коррекцию дефекта Т-клеток и частичное восстановление NK и В-клеточных линий [8]. У большинства пролеченных детей наблюдались признаки полного восстановление иммунной системы: нормальный ответ на стандартные для детского возраста прививки, излечение от инфекций и значительная прибавка в росте. Однако у 5 из 20 пациентов (в двух отдельных исследованиях) в результате векторного инсерционного онкогенеза со временем развился Т-клеточный лейкоз [5, 9].
Примерно в то же время исследовательская группа в Милане продемонстрировала значительный успех в исследованиях схожего редкого нарушения иммунодефицита, ADA-SCID [10]. Это заболевание, вызванное мутациями в гене аденозиндезаминазы (ADA), характеризуется отсутствием клеточного и гуморального иммунитета, повреждением органов, вызванным накоплением токсических метаболитов, и, как правило, летальным исходом на очень ранних этапах жизни. Хотя в первоначальные исследования не выявили терапевтического эффекта, последующие испытания в начале 2000-х годов оказались успешными для участвовавших в них двух пациентов. К такому успеху привели инновации, разработанные для обеспечения преимущественного выживания и приживления клеток, трансдуцированных ретровирусным вектором, содержащим ген ADA. Выживанию способствовало прерывание заместительной ферментной терапии ко времени реинфузии трансдуцированных клеток, а приживлению — мягкий режим кондиционирования перед введением генно-модифицированных клеток (кондиционирование — подготовка больного к пересадке ГСК с помощью цитостатической (химио- и лучевой) терапии — прим.переводчика) [10]. В 2009 году отчет о долгосрочном исходе у этих двух детей и восьми других пациентов показал безопасность, эффективность и долгосрочность генной терапии ГСК в лечении SCID у пациентов с дефицитом ADA, а также явное улучшение состояния пациентов и ослабление симптомов заболевания [11]. В 2016 году этот метод лечения получил одобрение EMA, что стало знаковым событием для сферы генной терапии. Для пациентов, у которых есть родной сибс с идентичным лейкоцитарным антигеном (HLA), трансплантация костного мозга по-прежнему остается терапией первой линии, однако у большинства пациентов таких сибсов не имеется. Для тех больных, у кого нет HLA-идентичного донора, эффективность генной терапии сопоставима с заместительной ферментной терапией, не требует повторных инъекций и не несёт риск нейтрализации антител к используемому ферменту крупного рогатого скота [12].
Интересно, что случаи инсерционного мутагенеза, характерные для исследований X-сцепленного SCID, никогда не наблюдались в исследовании ADA-SCID, хотя детальные исследования сайтов интеграции выявили небольшое различие между участками геномной интеграции в двух исследованиях [13-15]. Это привело к предположению, что наблюдаемые эффекты могут быть обусловлены другими факторами, включая сам продукт трансгена [16]. Исследования SCID подтвердили гипотезу о том, что перенос гена в ГСК можно использовать для лечения любого генетического заболевания, поддающегося терапии путем аллогенной трансплантации костного мозга. Более того, использование генетически модифицированных аутологичных клеток имело ряд преимуществ: отсутствие риска развития реакции «трансплантат против хозяина», гарантированная доступность «донора» (если только заболевание не поражает популяцию стволовых клеток пациента) и низкая вероятность неудачи приживления.
Как и в случае SCID-X1, в клинических испытаниях γ-ретровирусов наблюдались несколько случаев злокачественной трансформации, связанной с инсерционным онкогенезом, у пациентов с синдромом Вискотта — Олдрича [17] и Х-сцепленной хронической гранулематозной болезнью [18]. Исследования на склонных к опухолям модельных животных [19] и имеющиеся клинические данные показывают меньшую генотоксичность лентивирусных векторов (другой тип интегрирующих векторов) по сравнению с ретровирусными. Это частично связано с различиями в паттернах интеграции, поскольку γ-ретровирусные векторы склонны встраиваться вблизи точки старта транскрипционных единиц, тогда как лентивирусные векторы склонны встраиваться в транскрипционную единицу случайным образом, но не перед точкой старта транскрипции [20]. Важно отметить, что у лентивирусных векторов, применяемых клинически, удалены все их вирусные регуляторные элементы. Две вышеописанные характеристики обеспечивают более низкий риск активации соседних генов.
Подходы ex vivo с использованием лентивирусных векторов могут обеспечить долговременную стабильную экспрессию трансгена в центральной нервной системе (ЦНС). Первое испытание лентивирусной векторной трансдукции ГСК при нейродегенеративном расстройстве, Х-сцепленной адренолейкодистрофии (X-linked adrenoleukodystrophy, ALD) провели Cartier с соавт. [21]. Х-сцепленная ALD является смертельным демиелинизирующим поражением ЦНС, вызванным мутациями в гене ABCD1, кодирующем аденозинтрифосфат-связывающий кассетный транспортер. После введения лентивируса в аутологичные ГСК произошла поразительная стабилизация состояния больного, показавшая, что перенос генетического материала в стволовые клетки может работать как при нейродегенеративных, так и при иммунных нарушениях [21]. Этот корректирующий эффект основан на экспрессии донорского гена в микроглии, происходящей из циркулирующих моноцитов, которые перемещаются в мозг. Основываясь на данном наблюдении, исследователи продвинулись на шаг вперед и разработали лечение метахроматической лейкодистрофии, другого нейродегенеративного расстройства, которое ранее плохо поддавалось терапии трансплантацией костного мозга. Метахроматическая лейкодистрофия является лизосомальной болезнью накопления, вызванной мутациями в генеарилсульфатазе A (ARSA). Поздняя инфантильная форма заболевания характеризуется прогрессирующими двигательными и когнитивными нарушениями из-за накопления сульфатида — субстрата ARSA — в олигодендроцитах, микроглии и некоторых нейронах. Смерть наступает в течение нескольких лет с момента начала заболевания. Генная терапия с введением аутологичных ГСК до начала симптомов детям, родившимся с этим заболеванием, способствовала сохранению и дальнейшему приобретению основных двигательных и когнитивных навыков. Эффект терапии наблюдался в течение как минимум 32 месяцев после того, как пораженные сибсы пациентов начали утрачивать приобретенные навыки [22].
Более обширного переноса генетического материала в ГСК для достижения терапевтического эффекта требует терапия β-гемоглобинопатий. Это наиболее распространенные моногенные нарушения, причем наибольшей частотой встречаемости обладают β-талассемия и серповидноклеточная анемия (СКА) [23]. Учитывая нехватку HLA-идентичных доноров, разработка успешных методов генной терапии для коррекции аутологичных ГСК стала бы крупным прорывом, который мог бы принести пользу большому количеству пациентов во всем мире. Одна из основных проблем генной терапии β-гемоглобинопатий заключается в том, что клетки, несущие скорректированный ген, не имеют селективного преимущества перед больными клетками. Кроме того, экспрессия функционального глобина должна протекать специфическим для конкретного типа клеток образом и на уровне, значительном для оказания терапевтического эффекта (рассмотрено в источнике 24). Причиной β-талассемии являются мутации потери функции в гене β-глобина, приводящие к гемолитической анемии. В зависимости от тяжести, талассемию классифицируют как большую, промежуточную или малую. Пациентам с большой β-талассемией требуются постоянные переливания крови для лечения тяжелой анемии и хелатирование железа для предотвращения возникающего избытка железа. Причиной СКА, напротив, является замена аминокислоты, что приводит к дефекту пептида β-глобина, из-за чего эритроциты приобретают жесткость и серповидную форму, блокируют микрососуды и имеют меньшую продолжительность жизни. Несмотря на значительные успехи в лечении СКА, распространенность этого хронического заболевания даже в развитых странах остается высокой, а продолжительность жизни больных — сниженной [25].
Первый успешный метод генной терапии для взрослых с β-талассемией был опубликован в 2010 году [26]. На основании предыдущих наблюдений, перед реинфузией трансдуцированных лентивирусом аутологичных ГСК было проведено миелоаблативное кондиционирование, что способствовало стабильной выработке гемоглобина и отсутствию потребности в переливании в течение 33 месяцев. Следует отметить, что большую часть терапевтического эффекта обеспечил доминантный клеточный клон, в котором интегрированный вектор вызвал транскрипционную активацию HMGA2. Это доброкачественное клональное доминирование ослабло через несколько лет [24]. В настоящее время во многих регионах и странах проводятся спонсируемые промышленностью и некоммерческими организациями клинические испытания лентивирусных векторов для «исправления» β-глобина (NCT03276455, NCT03207009, NCT01639690, NCT02453477, NCT02151526). В недавнем отчете показано отсутствие необходимости в переливании крови после переноса гена у 12 из 13 пациентов с β-талассемией с частично функционирующим β-глобином (т.е. не с генотипом β0/β0). В тех случаях, когда экспрессия β-глобина полностью отсутствовала (т.е. при генотипе β0/β0), снижался средний объем крови, переливаемой за год, однако полностью прекратить переливания эритроцитов можно было только для 3 из 9 пациентов [27]. Следует отметить, что в этом исследовании не наблюдалось ни серьезных побочных эффектов, связанных с лекарственным препаратом, ни клонального доминирования, связанного с интеграцией векторов.
Существует лишь один отчет о применении лентивирусного подхода для лечения СКА [28]. ГСК от 13-летнего пациента обрабатывали лентивирусным вектором, несущим модифицированный против серповидноклеточности ген β-глобина с вариантом [βA87Thr:Gln (βA-T87Q)], и трансплантировали клетки обратно пациенту после миелоабляции. Через 15 месяцев после лечения уровень терапевтического β-глобина с модификацией против серповидноклеточности оставался на уровне ∼50% от всех β-подобных глобиновых цепей, что предотвращало рецидив кризов СКА и корректировало биологические признаки заболевания. Для определения возможности более широкого применения этого подхода необходимы более объемные группы пациентов и более длительное наблюдение.
После двух десятилетий наблюдений — с тех пор, как первых пациентов лечили клетками, модифицированными с использованием интегрирующих векторов — общие результаты клинических исследований подтверждают более высокую безопасность лентивирусных векторов по сравнению с γ-ретровирусными. Тем не менее, эти подходы требуют сложных процедур и обученного персонала для сбора, трансдукции и реинфузии гемопоэтических клеток-мишеней. Разработка и изготовление лентивирусных векторных систем также являются сложной задачей. Все эти факторы ограничивают число центров, которые могут сделать эти методы лечения доступными для пациентов. Например, Strimvelis® (терапия ADA-SCID) в настоящее время предлагается лишь в одном центре в Милане.
Генная терапия для лечения рака
В большинстве случаев клинические исследования переноса генов проводились при участии больных раком (Рисунок 1). В отличие от исследований генетических заболеваний, где критерии выбора трансгена однозначны, в исследованиях генной терапии рака применяли целый ряд стратегий для воздействия на опухоль. Некоторые из самых ранних исследований генной терапии рака были сосредоточены на целенаправленной доставке неактивной формы лекарства или гена «самоубийства», которые бы повышали чувствительность опухолевых клеток к цитотоксическим препаратам. Часто использовалась такая стратегия, как внутриопухолевая инъекция аденовирусного вектора, экспрессирующего ген тимидинкиназы (ТК). Клетки, поглощающие и экспрессирующие TK, можно убить введением ганцикловира (препарата, обычно применяемого для лечения герпесвирусных инфекций), который с помощью TK фосфорилируется до токсичного нуклеозида [29]. Аналогичный подход включает инъекцию аденовирусного вектора, который экспрессирует клеточный опухолевый антиген p53 (Ad-p53), приводящий к остановке роста и запуску апоптоза в опухолевых клетках. Введение Ad-p53 стало первой одобренной генной терапией, хотя на данный момент в США или Европе она не доступна. Эти ранние исследования проверили и подтвердили принцип действия генной терапии рака, но ясно показали, что серьезным препятствием для достижения полной эффективности выступает количество клеток, на которые должна воздействовать терапия, даже в том случае, когда вектор можно многократно вводить непосредственно в опухоли [30].
Другой подход заключается в использовании онколитических вирусов (OV), которые избирательно реплицируются в опухолевых клетках, не нанося вреда нормальным клеткам. Реплицирующийся вирус может размножаться в пределах опухоли, способствуя излечению. В данном обзоре OV подробно не рассматриваются, но недавние клинические испытания с нативными и генетически модифицированными вирусами подтвердили значительную клиническую пользу и малые риски для пациентов [рассмотрено в 31]. Первым OV, получившим в 2015 одобрение FDA в качестве противоопухолевой терапии (для лечения меланомы) в США стал талимоген лагерпарепвек (IMLYGIC®), модифицированный онколитический вирус герпеса, кодирующий гранулоцитарно-макрофагальный колониестимулирующий фактор [32].
Поскольку основной причиной смертности при большинстве типов рака является метастазирование, а не неконтролируемый рост первичной опухоли, значительный интерес представляет системная генная терапия. Новый класс терапевтических средств, продемонстрировавший обнадеживающий успех в лечении некоторых типов рака представляют генетически модифицированные иммунные клетки, экспрессирующие либо химерные антигенные рецепторы (chimeric antigen receptors, CAR), либо T-клеточные рецепторы (T-cell receptors, TCR), выделенные из опухолереактивных T-клеток. CAR придают генетически модифицированной клетке, обычно Т-клетке, новую специфичность к антигенам-мишеням, экспрессируемым на поверхности опухолевых клеток. Обычно CAR формируется путем связывания одноцепочечного вариабельного фрагмента моноклонального антитела с внутриклеточными сигнальными цепями, такими как домен CD3ζ, возникающий из TCR, и одним или несколькими внутриклеточными костимуляторными доменами. Такая архитектура позволяет обойти необходимость распознавания HLA, тем самым расширяя физиологический репертуар Т-клеток, например, до аутоантигенов, экспрессируемых в трансформированных клетках. С другой стороны, T-клетки со сконструированным TCR могут распознавать внутриклеточные антигены, не экспрессируемые на поверхности опухолевых клеток, до тех пор, пока они представлены HLA. Оба метода лечения включают получение Т-клеток от пациента путём афереза (переливания пациенту его собственной крови, из которой предварительно удалены определённые клеточные и плазменные составляющие — прим. переводчика), перенос генов (обычно с применением ретро- или лентивирусных векторных систем), экспансию Т-клеток ex vivo и кондиционирование, проводимое пациенту перед переносом клеток обратно (Рис.3).
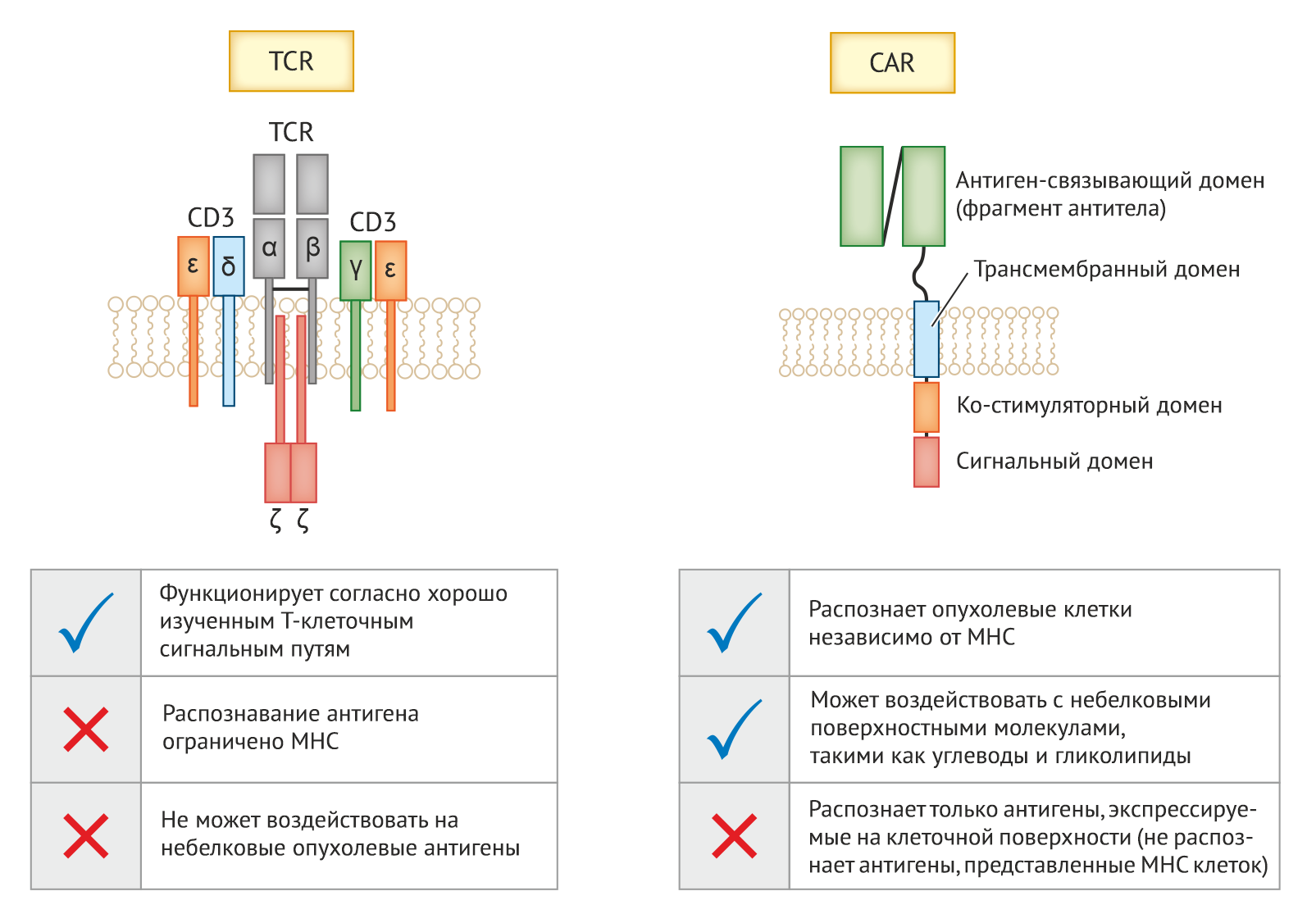
Сокращения: CAR — химерный антигенный рецептор; CD3 — кластер дифференцировки 3; MHC — главный комплекс гистосовместимости; TCR — T-клеточный рецептор.
В первых рядах CAR-технологий находится терапия, нацеленная на CD19, антиген, присутствующий в большинстве злокачественных новообразований В-клеточного происхождения, но отсутствующий в нормальных тканях, отличных от линии В-клеток. Переломный момент произошел в 2010 и 2011 годах, когда были опубликованы положительные результаты трех исследований CAR-терапии [33–35]. Эти исследования показали беспрецедентную противоопухолевую активность у пациентов с В-клеточной лимфомой [36, 37], хроническим лимфоцитарным лейкозом (NCT01029366, NCT00466531) или В-клеточным острым лимфобластным лейкозом [38]. Менее чем за 10 лет CAR T-клеточная иммунотерапия перешла от доклинического подтверждения механизма действия к нескольким успешным испытаниям терапии злокачественных новообразований В-клеточного происхождения, включая множественную миелому (NCT02135406) [39]. Эти успехи способствовали недавнему одобрению FDA и EMA двух методов CAR T-клеточной терапии, направленной на В-клетки, экспрессирующие CD19. Как уже упоминалось во введении, и KYMRIAH®, и и YESCARTA® показаны для лечения взрослых пациентов с рецидивирующей или рефрактерной В-клеточной крупноклеточной лимфомой, а KYMRIAH® также показан для лечения рецидивирующей и рефрактерной B-клеточной острой лимфобластной лейкемии у детей и молодых пациентов. Хотя CAR, нацеленные на CD19, остаются эталоном для всех других методов CAR-терапии, исследования CAR не ограничены воздействием на CD19 и включают терапию других гематологических злокачественных новообразований [рассмотрено в 31] и солидных опухолей [41]. Первое клиническое испытание с использованием Т-клеток с «искусственным» TCR было описано в 2006 году и показало объективную регрессию метастазов меланомы у 2 из 17 пациентов после адоптивного переноса лимфоцитов, нацеленных на MART-1 [42]. Успешность исследования открыла путь для серии исследований с воздействием на MART-1 и другие антигены меланомы. Хотя TCR-модифицированные клетки использовались для лечения различных типов рака [рассмотрено в 43], возможно, наиболее обнадеживающие клинические результаты наблюдались для лечения множественной миеломы, где частота ответа на терапию достигала 80% [44].
К сожалению, эти перспективные методы лечения имеют определенные риски. На сегодняшний день наиболее распространенные побочные эффекты после инфузии Т-клеток с CAR обусловлены активацией Т-клеток при взаимодействии с мишенями, к чему относят синдром высвобождения цитокинов, синдром активации макрофагов и синдром лизиса опухоли [45]. Такие токсические явления часто непродолжительны и устраняются с помощью поддерживающей терапии. Также в исследованиях с использованием генно-инженерных Т-клеток наблюдались серьезные побочные явления, в т.ч. со смертельным исходом, которые зачастую были связаны с атакой на клетки с другим типом антигена [46] или неопухолевые клетки со сходным типом антигена [47]. Контроль продолжительности воздействия и введение «защитного выключателя» может уменьшить риск побочных явлений. Хотя генная терапия при некоторых специфических опухолях произвела революционный эффект, необходимы дополнительные исследования, чтобы лучше понять механизмы, лежащие в их основе, и добиться лучшего контроля побочных реакций.
Несмотря на существующие ограничения, такие как рецидивы из-за того, что опухолевые клетки перестают экспрессировать антигены, являющиеся мишенью для CAR [48], генная терапия для лечения рака (в виде единичных или мультимодальных схем), по-видимому, близка к тому, чтобы стать новым поколением в терапии и улучшить исходы лечения онкологических больных [49]
Генная терапия in vivo
Генная терапия сетчатки
В случае наследственных дистрофий сетчатки, для которых не было фармакологической терапии, потенциальное лечение обеспечил AAV-опосредованный перенос генов. Заболевание обусловлено аутосомно-рецессивной мутацией в гене, кодирующем белок массой 65 кДа, связанный с пигментным эпителием (RPE65). Перенос этого гена, возможно, является первым клиническим подтверждением механизма действия любой генной терапии in vivo. Три независимых исследования (NCT00481546, NCT00516477 и NCT00643747) продемонстрировали улучшение зрительной функции у нескольких молодых пациентов в первые месяцы после одностороннего субретинального введения AAV2 [50–52]. Однако, только одно из этих исследований продвинулось до инъекции препарата в другой глаз и дошло до фазы III [53, 54]. Впоследствии в других исследованиях сообщалось, что у некоторых или всех изученных пациентов клинический эффект терапии, достигнутый вначале, постепенно ослабевал и оказался утрачен спустя 1-3 года после введения AAV (NCT00481546 и NCT00643747) [55, 56]. Такое явление не наблюдалось в исследовании, дошедшем до фазы III [54]. Во всех трех исследованиях использовался один и тот же серотип капсида AAV2 и конфигурация одноцепочечного генома кДНК RPE65. Точные причины различий в результатах являются предметом дискуссий, но, вероятно, они связаны с различиями в дизайне кассеты экспрессии, окончательной сборке векторной системы, хирургическом вмешательстве и иммуномодулирующем режиме в ходе операции. Эти переменные могут влиять на количество вектора, поступившего к клеткам-мишеням, тип и/или уровень потенциального иммунного ответа, степень подавления трансгенов или другие параметры, влияющие на клинический исход. Гипотеза о том, что субъекты на более ранней стадии заболевания будут иметь большую популяцию клеток-мишеней, которые могли бы поддерживать активность донорского гена, побудила Maguire с соавт. [57] расширить охват их исследования на фазе I, включив в нее детей. После обнадеживающих результатов этих исследований вышеупомянутые авторы продолжили оценку возможности дальнейшего улучшения функционального зрения, путем введения AAV2-RPE65 в другой, ранее лучше видящий глаз (NCT01208389) [53, 58]. В целом, наблюдалось улучшение показателей эффективности без значительной иммуногенности, что заложило основу для перехода клинического испытания в фазу III (NCT00999609) [54]. Фаза III рандомизированного контролируемого исследования показала превосходную эффективность препарата в группе испытуемых по сравнению с контрольной группой в первичной и первых двух вторичных конечных точках, а также некоторую положительную динамику в третьей вторичной конечной точке [54]. В 2017 году эти усилия завершились одобрением FDA препарата LUXTURNA® для лечения пациентов с дистрофией сетчатки, связанной с биаллельной мутацией RPE65. Это первая генная терапия для лечения генетического заболевания, одобренная в США.
Ранние успехи генной терапии аутосомно-рецессивных мутаций RPE65 запустили последующие исследования методов лечения других наследственных дистрофий сетчатки. В настоящее время проводятся исследования с использованием AAV-векторов для лечения наследственной оптической нейропатии Лебера (NCT02161380, NCT02652767), хороидеремии (NCT02341807, NCT02407678), Х-сцепленного ретиношизиса (NCT02317887, NCT02416622), Х-сцепленного пигментного ретинита (NCT03252847) и ахроматопсии (NCT02935517, NCT02599922, NCT03001310). Кроме того, в стадии разработки находятся исследования с использованием лентивирусных векторов, воздействующих на фоторецепторы, для лечения болезни Штаргардта (дегенерации жёлтого пятна) (NCT01367444) и синдрома Ашера типа 1B (NCT01505062). В дополнение к генетическим заболеваниям, методы генной терапии изучаются для лечения возрастной макулярной дегенерации (NCT01024998, NCT01301443, NCT01494805, NCT03066258).
Генная терапия, направленная на печень
Большая часть клинического опыта в области генной терапии, направленной на печень, основана на исследованиях по лечению гемофилии В с использованием AAV-векторов. Гемофилия — X-сцепленное нарушение свертывания крови, обусловленное мутациями в генах, кодирующих фактор VIII (приводит к гемофилии A) или фактор IX (приводит к гемофилии B). Заболевание характеризуется рецидивирующими кровотечениями и кровоизлияниями, в первую очередь, в суставы и мягкие ткани. В настоящее время гемофилию лечат путем внутривенной инфузии концентратов факторов свертывания, которые можно вводить либо профилактически, либо при эпизодах кровотечения.
В генной терапии гемофилии, направленной на печень, преимущественно используются AAV-векторы, воздействующие на гепатоциты, которые могут использоваться в качестве фабрик белка для выделения продукта трансгена в системное кровообращение. В первом исследовании терапии гемофилии B с помощью направленных на печень AAV семи пациентам через печеночную артерию вводили одноцепочечный AAV2-вектор, экспрессирующий фактор IX человека [7]. Первоначально эффективность наблюдалась у одного из двух пациентов, получивших самую высокую дозу вектора, 2×1012 векторных геномов (vg)/кг, с максимальным уровнем фактора IX, достигающим ~10% от нормы. Неожиданно примерно на 4 неделе после введения вектора началось бессимптомное самоограниченное повышение уровня печеночных трансаминаз, что совпало с началом постепенной потери активности фактора IX. В конечном итоге оба события были объяснены разрушением трансдуцированных гепатоцитов CD8+ T-клетками памяти со специфичностью к капсиду AAV [59]. Ни одно из доклинических исследований не предсказывало наблюдаемый иммунный ответ на капсид, и даже после десятилетия интенсивной работы этот феномен остается недостаточно изученным и плохо моделируемым на животных [60]. Второй пациент из подгруппы с введением высокой дозы также послужил источником ценных сведений: в том случае, когда вектор поступает через системное кровообращение, антитела против AAV, даже при невысоких титрах, предотвращают успешный перенос генов в клетки печени.
Генная терапия для нарушений центральной нервной системы и нервно-мышечных расстройств
Несмотря на то, что доклинические данные показали возможность генной терапии на основе AAV для лечения нарушений ЦНС, число клинических исследований с обнадеживающими результатами относительно невелико. Отчасти это связано с анатомической и функциональной сложностью человеческого мозга, в том числе с наличием гематоэнцефалического барьера, который ограничивает биораспределение вектора в ЦНС. Даже при условии успешной доставки вектора (например, путем прямого интрапаренхимального, интратекального, интрацеребровентрикулярного или системного введения) сложно добиться того, чтобы трансген внедрился в необходимое количество клеток либо в определенном регионе, либо в ЦНС в целом, что требуется для достижения адекватной степени безопасного усиления или ослабления гена в пределах терапевтического окна.
Первые клинические исследования с использованием AAV-векторов для доставки генов в ЦНС человека проводились с целью излечения болезни Канавана [67], болезни Паркинсона [68] и позднего детского нейронального цероидного липофусциноза [69]. Во всех этих исследованиях использовались векторы на основе AAV2, а их доставка осуществлялась локально в определенные области мозга; экспрессия была ограничена областями, близкими к месту инъекции, что желательно при болезни Паркинсона, но не в случае других заболеваний. Новые серотипы и различные методы инфузии с целью улучшения распределения вектора рассматривались в исследованиях поражений ЦНС, при которых требуется широкое распределение вектора. В качестве более эффективного способа для достижения такого глобального распределения предлагается доставка AAV-векторов в спинномозговую жидкость путем интрацеребровентрикулярного или интратекального введения [70].
Прорывное исследование Kaspar с соавт. продемонстрировало успешное воздействие на спинномозговые двигательные нейроны после системного введения AAV9 новорожденным мышам [71]. Впоследствии эта же группа ученых в исследовании I фазы продемонстрировала высокую эффективность лечения спинальной мышечной атрофии типа 1 (СМА1) у 15 пациентов путем аналогичного системного внутривенного введения AAV9 [72]. СМА1 является наиболее распространенной генетически обусловленной причиной смерти в младенческом возрасте: менее 20% больных доживают до 20 месяцев и не требуют искусственной вентиляции легких [72]. В изучаемой группе с высокими дозами терапии 12 пациентов продемонстрировали улучшение двигательных функций и выживаемости по сравнению с ожидаемым течением и естественной динамикой заболевания. В частности, 11 пациентов набрали более 40 баллов по шкале CHOP-INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) для количественной оценки двигательных навыков у детей с нервно-мышечными расстройствами, 11 контролировали положение головы, 8 сидели без посторонней помощи и 2 могли самостоятельно ползать, стоять и ходить. В целом, терапия способствовала достижению ключевых точек моторного развития, редко наблюдаемых у больных СМА1 [73]. В настоящее время исследование находится в фазе III (NCT03306277).
Некоторые проблемы в разработке успешной терапии, направленной на ЦНС, включают необходимость тщательного мониторинга потенциальных иммунных реакций, особенно против трансгенного продукта, и улучшения тропизма вектора для воздействия на большее количество клеток при более низких дозах, а также более точного воздействия на конкретные типы клеток в разных областях мозга. Эффективность важнейшего исследования СМА1, наряду с благоприятным профилем безопасности клинических испытаний, нацеленных на ЦНС, в более чем десятилетнем клиническом опыте с использованием AAV-векторов, говорят о необходимости дальнейшего развития этой технологии для лечения сложных многофакторных неврологических состояний, таких как болезни Паркинсона и Альцгеймера.
Редактирование генома
Данный обзор сфокусирован на усилительной генной терапии, в которой функциональный ген переносится в ткань-мишень для стимуляции экспрессии генного продукта, что оказывает терапевтический эффект (Рис.2).
Еще один привлекательный метод, находящийся на стадии разработки, — это редактирование генома, в котором мутация корректируется in situ, приводя к созданию в физиологически релевантной ткани-мишени копии гена дикого типа под контролем эндогенных регуляторных сигналов. Этот подход использует новые инструменты, в том числе нуклеазы цинковых пальцев, эффекторные нуклеазы, подобные активаторам транскрипции и CRISPR/Cas9 [рассмотрено в 74]. Это различные типы специфичных к последовательности нуклеаз, которые осуществляют двухцепочечные разрывы в ДНК рядом с местом мутации, а затем используют донорную последовательность (шаблон) и клеточные механизмы репарации ДНК (например, гомологически направленную репарацию) для восстановления или коррекции функционирующего гена. Редактирование генома также может быть использовано для подавления экспрессии генов путем формирования мутаций сдвига рамки считывания в месте разрыва ДНК. Тем не менее, только в трёх клинических исследованиях генной терапии in vivo используется нуклеаза — для лечения гемофилии B (NCT02695160), мукополисахаридоза типа I (NCT02702115) и мукополисахаридоза типа II (NCT03041324). В данных случаях нуклеаза применяется не с целью коррекции мутации, а, скорее, для целенаправленной интеграции донорских кассет AAV в участки двухцепочечного разрыва ДНК в локусе альбумина. Выбор альбумина обоснован высокой транскрипционной активностью его промотора и возможностью использования локуса как безопасного участка для интеграции трансгена [75]. Поскольку у взрослых периодичность обновления гепатоцитов относительно низка, и, следовательно, нет необходимости в интеграции векторов для достижения устойчивого уровня экспрессии после переноса гена [62], этот подход может найти лучшее применение в педиатрии. Однако, два основных способа применения редактирования генома ex vivo направлены на разрушение собственных пораженных генов больного, а не на интеграцию донорских последовательностей. В частности, нуклеазы цинковых пальцев [76] и CRISPR (NCT03164135) применялись для разрушения корецептора ВИЧ-1 CCR5 и лечения ВИЧ-инфекции. Метод CRISPR также набирает обороты в модификации Т-клеток для иммунотерапии рака (NCT02793856).
Заключение
Стратегии переноса генов настолько мощны и универсальны, что лишь для нескольких серьезных заболеваний пока не разрабатываются методы переноса генов. Разработка новых классов терапевтических средств обычно занимает два-три десятилетия (например, моноклональные антитела и рекомбинантные белки). Тот же путь прошла генная терапия, внедренная в клинические испытания в начале 1990-х годов. Примеров клинического успеха в настоящее время достаточно, и подходы генной терапии, вероятно, будут становиться все более важными. Центральным вопросом является долгосрочная безопасность переноса генов, поэтому регулирующие органы требуют в течение 15 лет наблюдать за пациентами, участвовавшими в испытаниях генной терапии. Реализация терапевтических преимуществ современной молекулярной медицины будет зависеть от постоянного прогресса в технологии передачи генов.
