
Калиевые каналы внутреннего выпрямления: часть 3

В первой и второй частях речь шла о калиевых каналах внутреннего выпрямления, которые участвуют в поддержании потенциала покоя, а также в трансэпителиальном и трансастроцитарном транспорте калия. Сейчас мы поговорим про калиевые каналы, активируемые G-белками, которые играют важную роль в передаче сигнала многих рецепторов гормонов и нейромедиаторов. Мутации в этих каналах приводят к нарушениям развития, первичному гиперальдостеронизму и удлинению интервала QT.
Kir3.x/GIRK – калиевые каналы, активируемые G-белками
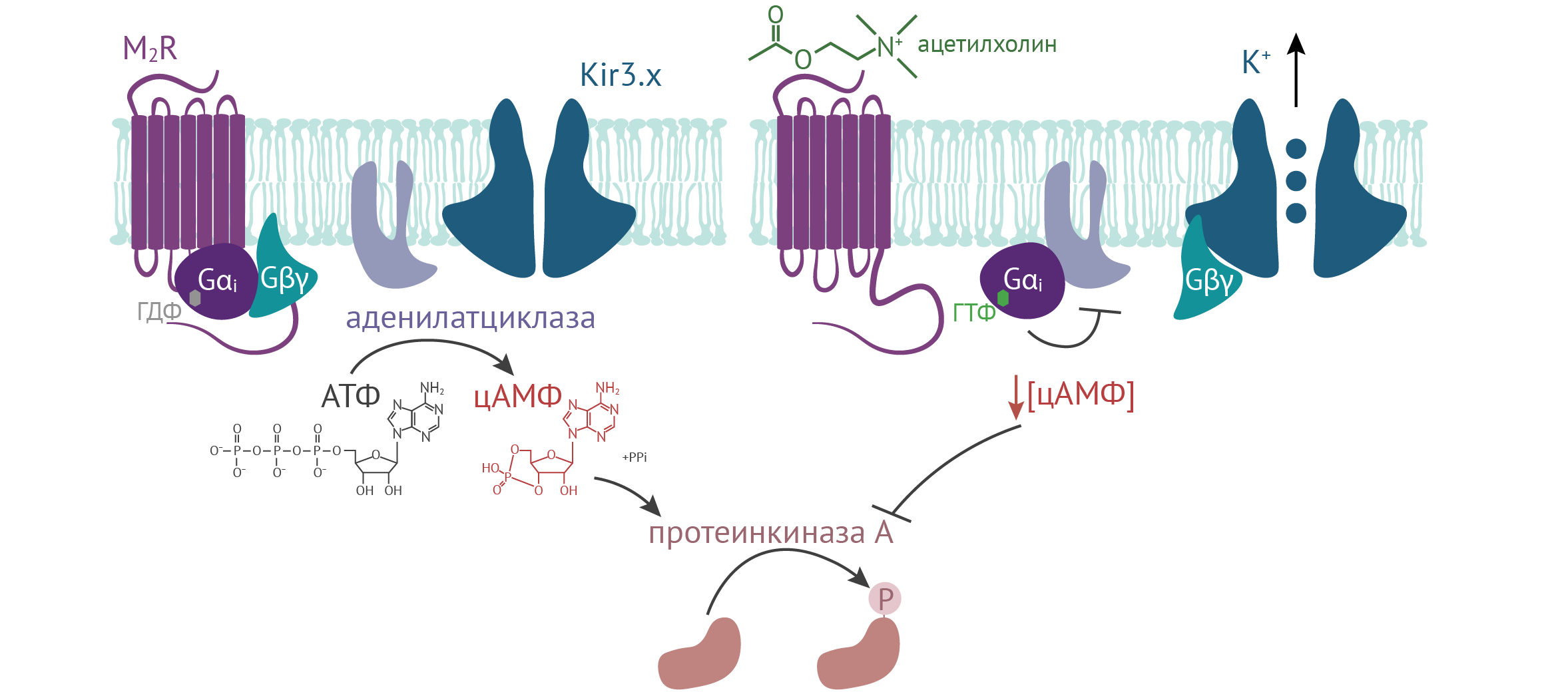
В геноме человека закодировано более 800 типов рецепторов, сопряженных с G-белками (GPCR — G protein-coupled receptors), лигандами которых обычно служат гормоны, нейромедиаторы, цитокины, одоранты и другие молекулы. Активация рецептора, в свою очередь, ведет к активации тримерного G-белка, субъединицы которого, Gɑ и Gβγ, запускают внутриклеточные сигнальные каскады. Субъединицы Gɑ подразделяются на три классических типа: стимулирующие аденилатциклазу Gɑs, ингибиторные Gɑi и стимулирующие фосфолипазу C Gɑq. Каналы подсемейства Kir3.x, также называемые GIRK1 (G protein-coupled inwardly-rectifying K+ channels), включены в каскад работы ряда GPCR и активируются Gβγ-субъединицами некоторых G-белков (см. Рисунок 18). Так, в сердце эти каналы вызывают гиперполяризацию мембраны, например, в ответ на стимуляцию ацетилхолином М2-холинорецепторов или аденозином А1-пуринергических рецепторов. В других возбудимых клетках GIRK участвуют в сигнальных каскадах Gi-сопряженных рецепторов соматостатина [1], рецепторов серотонина 5HT1 [2], GABAB, дофаминовых D2-рецепторов [3], ɑ2-адренорецепторов, δ-, κ- и μ-опиоидных рецепторов [4, 5] и др.
1 — Для этого подсемейства закрепилась старая номенклатура GIRK1–4 вместо унифицированной Kir3.1–4, и эти две системы существуют параллельно.
У человека GIRK представлены четырьмя изоформами, которые образуют гомотетрамеры и гетеротетрамеры различного состава; функциональные тетрамерные комплексы также называют KG-каналами.
Первым был клонирован GIRK1/Kir3.1/KCNJ3 из библиотеки кДНК сердца крысы. Было показано, что Kir3.1 входит в состав KG-каналов [6, 7]. Однако гомотетрамеры Kir3.1 нефункциональны [8–11], поскольку удерживаются в ЭПР [12, 13]. Kir3.1 на плазматической мембране существуют в составе комплексов Kir3.1/3.2 [14] или Kir3.1/3.4 [8].
Kir3.2/GIRK2/KCNJ6 представлен несколькими сплайс-вариантами [15–17] с тканеспецифичными паттернами экспрессии, которые могут образовывать как гомомеры [15, 18], так и гетеромеры [18–21]. Гомомеры Kir3.2 находятся на плазматической мембране а также на везикулах в цитоплазме, поскольку на их N-конце есть мотив, запускающий эндоцитоз канала [12, 22].
Два варианта Kir3.3/GIRK3/KCNJ9 были обнаружены в библиотеке кДНК мозга мыши [14, 23], однако результаты исследования свойств данных изоформ противоречивы: в части работ коэкспрессия Kir3.3 с Kir3.1 [12, 24, 25], Kir3.2 [24, 25] или Kir3.4 [25] не давала измеримых токов, в других удавалось зарегистрировать высокоамплитудные токи гетеромеров Kir3.3/Kir3.1 [9, 23, 26] и Kir3.3/Kir3.2 [26, 27]. Коэкспрессия Kir3.3 с Kir3.2, Kir3.1/3.2 Kir3.1/3.4 в общем случае приводит к снижению токов, поскольку Kir3.3 не имеет сигнала экспорта из ЭПР, но несет сигнал лизосомной локализации [12, 24, 25]. На C-конце изоформ Kir3.3 и Kir3.2c находится PDZ-связывающий мотив (postsynaptic density-95, discs large, zona occludens domain), который важен для локализации на плазматической мембране, осуществляемой через взаимодействие с PDZ-доменом белка SNX27 (Sorting nexin 27) [26].
Kir3.4/GIRK4/CIR/KCNJ5 был обнаружен в ткани предсердия крысы методом иммунопреципитации с антителом против Kir3.1 (при этом в осадок выпадает не только белок, против которого нацелено антитело, но и все другие компоненты комплекса, в который входит этот белок) [28]. Различными методами было показано, что гетеромеры Kir3.1/Kir3.4 образуют сердечные KG-каналы [28–31], однако в предсердиях также были обнаружены гомотетрамеры Kir3.4 [32].
Регуляция G-белками, PIP2 и [Na+]i
Субъединицы Gβγ, ассоциированные с различными рецепторами, различаются способностью активировать каналы GIRK, причем эта селективность зависит именно от рецептора и не зависит от конкретных подтипов Gβ и Gγ. Удивительно, что человеческий рецептор может стимулировать KG-ток в клетках кукурузной листовой совки Spodoptera frugiperda (клетки этих бабочек обычно используются для синтеза рекомбинантных белков) с помощью эндогенных Gβγ [33]. Так, калиевый ток стимулируют Gβγ, связанные с Gɑi-сопряженным M2R, но не с Gɑs-сопряженным β1- и β2-адренергическими рецепторами (β1AR и β2AR) и не с Gɑq-сопряженными рецепторами. До недавних пор селективность Gβγ-субъединиц оставалась загадкой. Если активация Gɑq снижает содержание PIP2 за счет гидролиза последнего фосфолипазой C, и недостаточный уровень PIP2 препятствует активации канала [34], то вопрос о селективности между Gɑi и Gɑs оказался несколько сложнее. Высказывались гипотезы о существовании GIRK в виде макромолекулярного комплекса с Gɑi-, но не с Gɑs-сопряженными рецепторами [35, 36], или о более сильном связывании Gβγ с комплексом Gɑs • ГТФ, из-за чего количества Gβγ-субъединиц оказывается недостаточно для активации GIRK [37]. В серии убедительных экспериментов Тухара и МакКиннон2 показали, что Gɑi-сопряженные рецепторы высвобождают Gβγ быстрее и в большем количестве, чем рецепторы, сопряженные с Gɑs, и эти Gβγ способны влиять не только на GIRK, но и на другие мишени (например, ингибировать TRPM3) [38]. Таким образом, специфичность субъединиц Gβγ определяется кинетическими параметрами.
2 — Родерик Маккиннон стал лауреатом Нобелевской премия по химии совместно с Питером Агре за открытие трехмерной молекулярной структуры бактериального калиевого канала и исследование природы его селективности.
Каналы, содержащие субъединицы Kir3.2 или Kir3.4, могут активироваться при повышении концентрации Na+ в цитозоле с EC50 ~ 30–40 мМ [25, 39, 40]. Этот механизм важен для ограничения возбудимости клеток: при продолжительной деполяризации мембраны излишний натрий активирует KG-ток, который возвращает мембранный потенциал к нормальному значению потенциала покоя. За чувствительность Kir3.2 к Na+ отвечает остаток аспартата D228, который находится в цитоплазматическом домене канала [39, 40].
Физиологическая роль калиевых каналов, активируемых G-белками
Kir3.1/3.4 в сердце
Выделение ацетилхолина терминалями блуждающего нерва активирует Gi-сопряженный M2-холинорецептор и снижает частоту сердечных сокращений. Помимо понижения концентрации цАМФ, этот эффект вызван усилением K+-проводимости через KG-каналы, также называемые KACh, в синоатриальном узле (см. Рисунок 18). Эти каналы состоят из субъединиц Kir3.1/3.4, и у нокаутных по Kir3.4 мышей K+-ток внутреннего выпрямления в ответ на стимуляцию ацетилхолином отсутствует [41]. В покое сердечный ритм у этих мышей был в норме [41] или несколько повышен [42], однако в ответ на парасимпатическую стимуляцию у них не наблюдалась брадикардия. Кроме того, у нокаутных мышей пропала вариабельность сердечного ритма, которая в норме отражает баланс между парасимпатическими и симпатическими влияниями [43].
Помимо ацетилхолина вызывать брадикардию может также аденозин, связываясь с A1-пуринергическими рецепторами (каскад аналогичен тому, что представлен на Рисунке 18) [44].
Kir3.x в мозге
KG-каналы в мозге расположены как на пресинаптических, так и на постсинаптических мембранах [45]. Открытие постсинаптических KG-каналов создает медленные (в отличие от быстрых, вызванных активацией ионотропных рецепторов) тормозные постсинаптические потенциалы (ТПСП) и снижает возбудимость постсинаптической клетки. В мозге представлен весь спектр известных гомо- и гетеромерных KG-каналов, однако различные комбинации распределены между разными отделами мозга и типами нейронов. Сложные и перекрывающиеся паттерны экспрессии субъединиц создают разнообразие функций KG-каналов в мозге, возможности для их тонкой регуляции и дополнительный уровень устойчивости к нарушениям. Kir3.1, Kir3.2 и Kir3.3 экспрессируются во многих областях мозга, а Kir3.4 отличается более узкой локализацией [45].
Данные, полученные на мышах с пониженной экспрессией Kir3.2, показали, что Kir3.2 участвует в генерации ТПСП в нейронах гиппокампа и мозжечка [46, 47], а у Kcnj6−/− мышей снижен судорожный порог [46].
Kcnj9−/− мыши не имеют значительных нарушений и кажутся здоровыми [48], однако Kir3.3 все же участвует в генерации ТПСП. Так, в нейронах голубого пятна присутствуют Kir3.1, Kir3.2 и Kir3.3 [19, 49]. В этих нейронах мет-энкефалин через μ-опиоидные рецепторы вызывает гиперполяризацию мембраны [50–52]. У Kcnj6−/− (нокаутных по Kir3.2) мышей гиперполяризация снижена на 40 % по сравнению с мышами дикого типа, а у двойного нокаута Kcnj6−/−/9−/− — на 80 % [48], остаточный ответ скорее всего опосредован цАМФ-зависимыми катионными каналами. Такие результаты можно объяснить присутствием гетеромеров Kir3.1/3.2 и Kir3.1/3.3, и при нокауте Kir3.2 или Kir3.3 оставшаяся изоформа компенсирует дефект, что свидетельствует в пользу существования Kir3.1/3.3 гетеромеров in vivo, которые в экспериментальных системах экспрессии нефункциональны [24, 25]. Похожие результаты были получены на нейронах зоны CA1 гиппокампа, в которых агонисты Gi-сопряженных GABAB-рецепторов (ГАМК и баклофен) вызывают ТПСП через KG-каналы [53].
Kir3.1, 3.2 и 3.3 в системе подкрепления
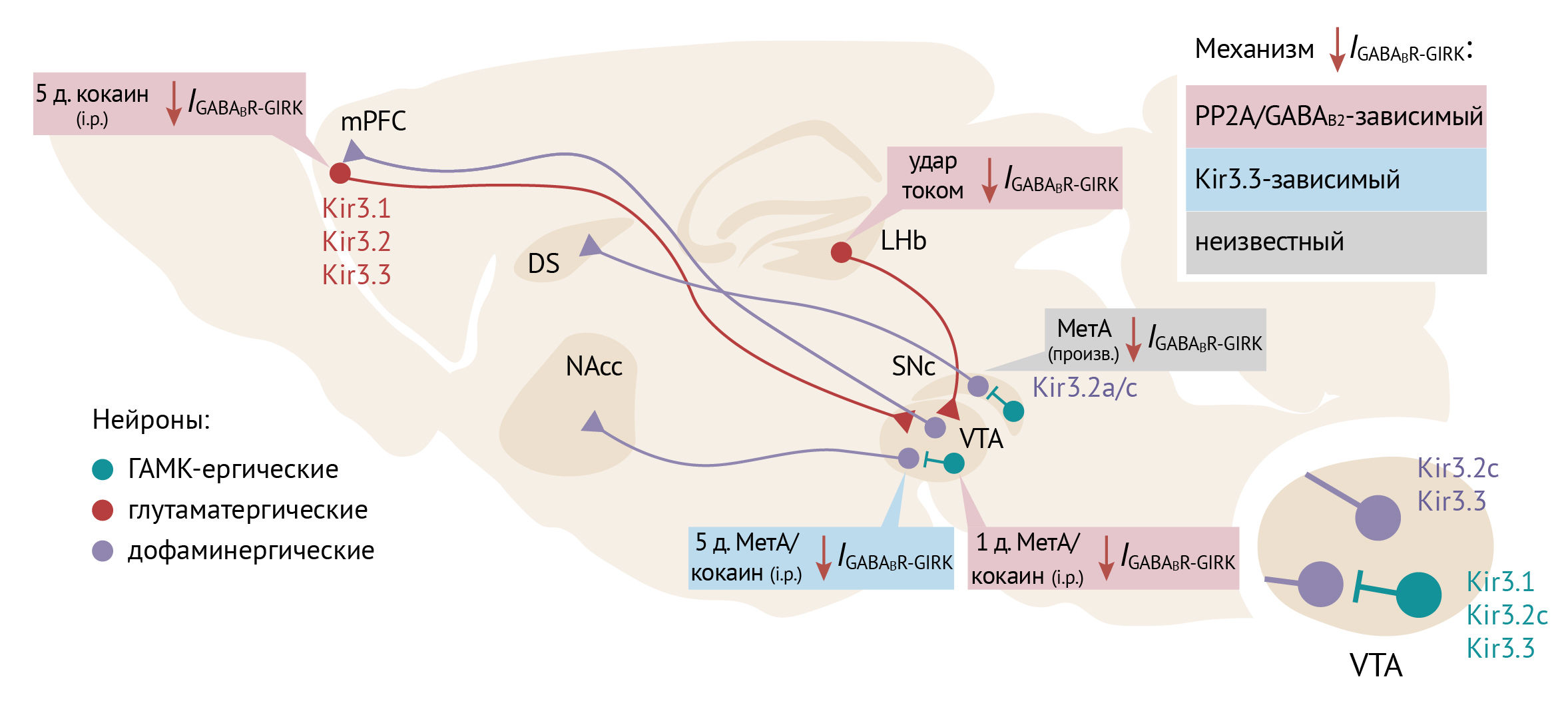
Вещества, вызывающие аддикции, захватывают механизмы синаптической пластичности, в нормальных условиях используемые организмом в различных адаптивных процессах. Ключевую роль в системе подкрепления играет вентральная область покрышки (VTA — ventral tegmental area), в которой есть дофаминергические и ГАМК-ергические нейроны. Дофаминергические проекции из VTA направляются в прилежащее ядро (NAcc), медиальную префронтальную кору (mPFC) и другие области, связанные с обучением и вознаграждением (так называемый мезолимбический путь). Терминали ГАМК-ергических нейронов оканчиваются в том числе на дендритах дофаминергических клеток, и их сигнал ингибирует высвобождение дофамина (см. Рисунок 19; на русском языке о «системе вознаграждения» можно почитать на сайте [54]). В системе подкрепления Kir3.2/Kir3.3 опосредует тормозящие эффекты GABAB и D2R и участвуют в процессах синаптической пластичности и развитии аддикций.
В дофаминергических нейронах VTA-токи через GIRK-каналы, вызванные стимуляцией GABAB-рецепторов (IGABABR-GIRK), чувствительны к характеру активности нейронов. Залповая активность (5 стимулов по 20 Гц, каждый 500 мс в течение пяти минут) ведет к усилению IGABABR-GIRK, а тоническая (2 Гц в течение 2,5 минут) снижает IGABABR-GIRK. Подобные вариации амплитуды медленных ТПСП свидетельствуют об изменении возбудимости нейрона в зависимости от паттерна его активности [55].
Эффекты веществ, вызывающих зависимость, сходятся на повышении уровня дофамина в VTA, NAcc и mPFC [56–58]. Так, опиоиды через μ-рецепторы тормозят секрецию ГАМК в ГАМК-ергических нейронах ростромедиального ядра покрышки и VTA, что приводит к снятию ингибирования с дофаминергических нейронов и усилению выделения дофамина [59, 60] (далее этот механизм обсуждается подробнее). Амфетамин и метамфетамин поступают в синаптические терминали и обращают транспорт дофамина из везикул в цитоплазму [61], кокаин блокирует обратный захват дофамина через DAT (дофаминовый транспортер, является к тому же маркером дофаминергических нейронов) в синапсах нейронов VTA [62, 63] и повышает уровень дофамина в областях мозга, получающих дофаминергические входы из VTA (например, NAcc), а также в самой VTA. Как в большинстве химических синапсов, здесь существует механизм отрицательной обратной связи, контролирующий высвобождение дофамина: это происходит путем активации D2-ауторецепторов на терминалях нейронов VTA. Аксоны ГАМК-ергических нейронов активируют GABABR на нейронах VTA, что в сумме ведет к ингибированию этих нейронов через каналы GIRK, связанные с Gi-сопряженными рецепторами [64]; это, в свою очередь, тормозит локомоторную активность. Хроническое воздействие кокаина ослабляет передачу сигнала через тормозные GPCR, тем самым вызывая сенсибилизацию, в развитии которой также участвуют каналы GIRK [65, 66].
Этанол повышает активность дофаминергических нейронов VTA через различные молекулярные мишени [67], в число которых входят и каналы GIRK. Было показано, что этанол способствует открытию GIRK независимо от G-белков [68].
Роль каналов Kir3.x в развитии аддикций изучают с помощью нокаутных моделей на грызунах. Результаты, полученные на тотальных нокаутах по отдельным субъединицам Kir3.x, свидетельствуют о сложных взаимодействиях этих субъединиц в различных структурах системы подкрепления. Одним из подходов к изучению эффектов психостимуляторов на животных служит измерение локомоторной сенсибилизации — усиления стимулирующего локомоторную активность эффекта кокаина и других психостимуляторов при каждом повторном применении. Грызунам вводят психостимулятор в течение нескольких дней и регистрируют общую дистанцию, которую животное прошло за период наблюдений. В норме на пятый день введения животные сенсибилизируются и проходят значительно большее расстояние, чем в первый день инъекций.
Когда этим методом исследовали Kcnj6−/− и Kcnj9−/− мышей, а также двойных нокаутов, у которых отсутствуют соответственно Kir3.2 и Kir3.3, выяснилось, что нокаут Kir3.2 усиливает локомоторную сенсибилизацию в ответ на инъекции кокаина, в то время как Kcnj9−/− мыши и мыши с двойным нокаутом Kcnj6−/−/9−/− по данному показателю не отличались от мышей дикого типа. В экспериментах с произвольным потреблением кокаина животные с одиночным нокаутом по Kir3.2 и Kir3.3 употребляли меньше, чем мыши дикого типа, а животные с двойным нокаутом употребляли количество кокаина, промежуточное между таковым у мышей с одиночным нокаутом и у мышей дикого типа [69].
В работе с нокаутами Kir3.1 и Kir3.2 авторы также обнаружили, что у нокаутных животных выявляется более сильная реакция на кокаин, чем у мышей дикого типа. Затем, предположив, что такие эффекты должны быть вызваны изменениями в дофаминергических нейронах VTA, авторы измерили IGABABR-GIRK (при стимуляции GABAB-рецепторов баклофеном) в этих нейронах у Kcnj3−/− мышей и не нашли отличий по сравнению с мышами дикого типа (что согласуется с отсутствием Kir3.1 в этих нейронах), в то время как у Kcnj6−/− мышей инъекции кокаина снижали IGABABR-GIRK [70]. Эти результаты свидетельствуют о том, что влияние KG-каналов на эффекты, вызываемые психостимуляторами, должно реализовываться не только в дофаминергических нейронах VTA, но и в других областях мозга [64].
Результаты, полученные на животных с тотальными нокаутами, не позволяют прояснить детали механизма модуляции эффектов психостимуляторов каналами Kir3.х. Примерно столь же сложны для интерпретации результаты исследования воздействия других аддиктивов. Поэтому исследователи переходят на тканеспецифичные нокаутные модели, основанные на системе Cre-lox. Эта система работает следующим образом: трансгенных мышей, у которых рекомбиназа Cre экспрессируется под контролем какого-либо промотора, скрещивают с мышами, у которых таргетный ген окружен сайтами loxP — специальными последовательностями ДНК, которые узнает и вырезает рекомбиназа Cre. Так можно вызвать нокаут таргетного гена в дофаминергических нейронах (DAT-Cre вырезает ген только в нейронах, экспрессирующих транспортер дофамина DAT, то есть в дофаминергических нейронах). Кроме этого, можно использовать аденовирусы, содержащие ген рекомбиназы Cre, и инъецировать их в определенные области мозга мышей. В этом случае можно ограничить удаление гена какой-то определенной небольшой областью мозга.
Нокаут Kir3.2 в дофаминергических нейронах, как и тотальный нокаут Kcnj6−/−, усиливает локомоторную сенсибилизацию в ответ на кокаин, а также увеличивает количество произвольно потребленного мышью кокаина [71]. Однако нокаут во всех дофаминергических нейронах не позволяет локализовать наблюдаемые эффекты, поскольку, например, дофаминергические нейроны компактной части черной субстанции не только регулируют локомоцию, но и участвуют в подкреплении [72, 73].
Локализация каналов Kir3.2c/3 на плазматической мембране находится под контролем белка SNX27 [26], и нокаут соответствующего гена в дофаминергических нейронах (при этом количество GIRK каналов на мембране снижается) ведет к усилению реакции на однократное введение кокаина [74], снижая торможение через GABABR. Локомоторная сенсибилизация в ответ на хроническое введение кокаина у этих мышей также усиливается. Гиперэкспрессия в этих нейронах GIRK2a, который не содержит PDZ-связывающего мотива и не чувствителен к SNX27, возвращает фенотип к норме [75]. Интернализация каналов GIRK в дофаминергических нейронах VTA под действием метамфетамина зависит от присутствия в составе этих каналов субъединицы Kir3.3 и не зависит от дефосфорилирования GABAB (см. ниже): у Kcnj9−/− мышей не наблюдалось снижения IGABABR-GIRK после введения метамфетамина в течение пяти дней. Авторы предполагают, что взаимодействие Kir3.2c/3 c SNX27 может быть опосредованно именно субъединицей Kir3.3 [76].
Таким образом, можно предложить модель, согласно которой при хроническом воздействии кокаина на мембране дофаминергических нейронов понижается количество каналов GIRK за счет снижения экспрессии SNX27 [77], уменьшается чувствительность нейронов к тормозным медиаторам и формируется сенсибилизация.
Психостимуляторы влияют и на каналы GIRK в других нейронах и отделах мозга (см. Рисунок 19). В ГАМК-ергических нейронах VTA кокаин и метамфетамин вызывали снижение IGABABR-GIRK путем интернализации GABAB1 и GIRK2, что происходило совместно с дефосфорилированием GABAB2 по остатку S783 (GABAB рецептор — это гетеродимер, состоящий из субъединиц GABAB1 и GABAB2; фосфорилирование GABAB2S783 способствует локализации рецепторного комплекса на плазматической мембране). Ингибирование фосфатазы PP2A окадаевой кислотой в остром эксперименте повысило IGABABR-GIRK до нормального уровня [78]. Снижение ингибирования в ГАМК-ергических нейронах VTA в ответ на психостимуляторы может служить адаптацией к повышенной возбудимости дофаминергических нейронов [79].
Нейроны LHb (латерального ядра поводка) активируются стимулами, вызывающими аверсию (отвращение), и участвуют в формировании абстинентного синдрома при прекращении приема кокаина [80]. У мышей, которых подвергали ударам тока (этот протокол вызывает симптомы депрессии), был снижен IGABABR-GIRK. Эти симптомы ослаблялись окадаевой кислотой, что может свидетельствовать в пользу того же механизма, который описан для ГАМК-ергических нейронов VTA [81].
Этанол
Каналы GIRK влияют на поведение, связанное с потреблением не только психостимуляторов, но и других аддиктивов, например, этанола. Kcnj6−/− мыши поглощали больше этанола по сравнению с контрольной группой мышей дикого типа; при этом у них была повышена активность в открытом поле. Этанол не оказывал на таких мышей столь сильного анксиолитического действия, как на мышей дикого типа, и симптомы отмены были ослаблены [85]. Kir3.3 снижает потребление алкоголя: нокаутные животные поглощали больше этанола, и у них был слабее выражен синдром отмены [86]. После инъекция вируса, содержащего ген Kcnj9, в VTA фенотип нокаутных мышей вернулся к нормальному [86].
Опиоиды
Ранее предполагали, что активация тока через KG-каналы при стимуляции μ-опиоидных рецепторов ГАМК-ергических нейронов в VTA и ростромедиальном ядре покрышки усиливает активность дофаминергических нейронов, устраняя их торможение [59, 60]. Однако результаты исследования, выполненного на GAD2-Cre+/−/Kcnj6lox/lox мышах, у которых отсутствует экспрессия Kir3.2 только в ГАМК-ергических нейронах, противоречат этому представлению. У нокаутных мышей повышение активности ГАМК-ергических нейронов не сказалось на локомоторной реакции на введение морфина (локомоторная сенсибилизация оценивалась так же, как и в описанных выше исследованиях эффектов психостимуляторов), в то время как нокаут в дофаминергических нейронах усилил локомоторный ответ на морфин [87]. Эти результаты говорят о том, что Kir3.2-содержащие каналы опосредуют эффекты морфина, модулируя активность дофаминергических, но не ГАМК-ергических нейронов, однако другие эффекты μ-опиоидных рецепторов могут оставаться в рамках канонического механизма.
Kir3.4 в гиппокампе и центрах регуляции голода и насыщения
Kir3.4 в гиппокампе экспрессируется слабо [88, 89], однако у Kcnj5−/− мышей нарушены пространственное обучение и память [88].
Особенно высока экспрессия Kir3.4 в вентромедиальном гипоталамусе, в вентромедиальном (VMN) и аркуатном ядрах [42]. В частности, гипоталамус участвует в регуляции энергообмена, и вентромедиальный гипоталамус считают «центром насыщения» [90]. VMN и POMC (проопиомеланокортиновые) нейроны аркуатного ядра — это основные нервные центры насыщения, стимулирующие трату энергии. И наоборот, орексиновые нейроны латерального гипоталамуса и экспрессирующие NPY (нейропептид Y) нейроны аркуатного ядра представляют собой «центр голода» и запускают потребление пищи [91] (подробнее см. про регуляцию пищевого поведения). Эти наблюдения послужили основой для гипотезы о роли Kir3.4 в регуляции энергообмена.
Действительно, нокаутные по Kir3.4 взрослые мыши набирали лишний вес за счет увеличения потребления пищи и снижения трат энергии при физической активности даже несмотря на тахикардию и увеличение температуры тела в покое, вызванные отсутствием KACh-каналов в сердце. Кроме того, нокаутные мыши лучше справлялись с задачами на инструментальное обучение с пищевым подкреплением — возможно, за счет модуляции активности нейронов в VTA Kir3.4-содержащими каналами, либо за счет проекций из VMN в VTA и прилежащее ядро [42].
Kir3.x в поджелудочной железе
Секреция инсулина и глюкагона островками Лангерганса не только зависит от концентрации глюкозы в плазме крови, но и модулируется различными нейромедиаторами и гормонами. Передача сигналов от некоторых рецепторов этих лигандов опосредована гиперполяризацией мембраны при открытии KG-каналов. Так, в ɑ- и в β-клетках экспрессируются Kir3.2c и Kir3.4 [92–94], гомо- и гетеромерные сочетания которых образуют KG-каналы поджелудочной железы. Катехоламины через ɑ2-адренорецепторы ингибируют секрецию инсулина β-клетками, а соматостатин тормозит секрецию как инсулина, так и глюкагона [95–98].
Мутации Kir3.x/GIRK
Мутации Kir3.2/GIRK2: синдром Кеппена-Любински
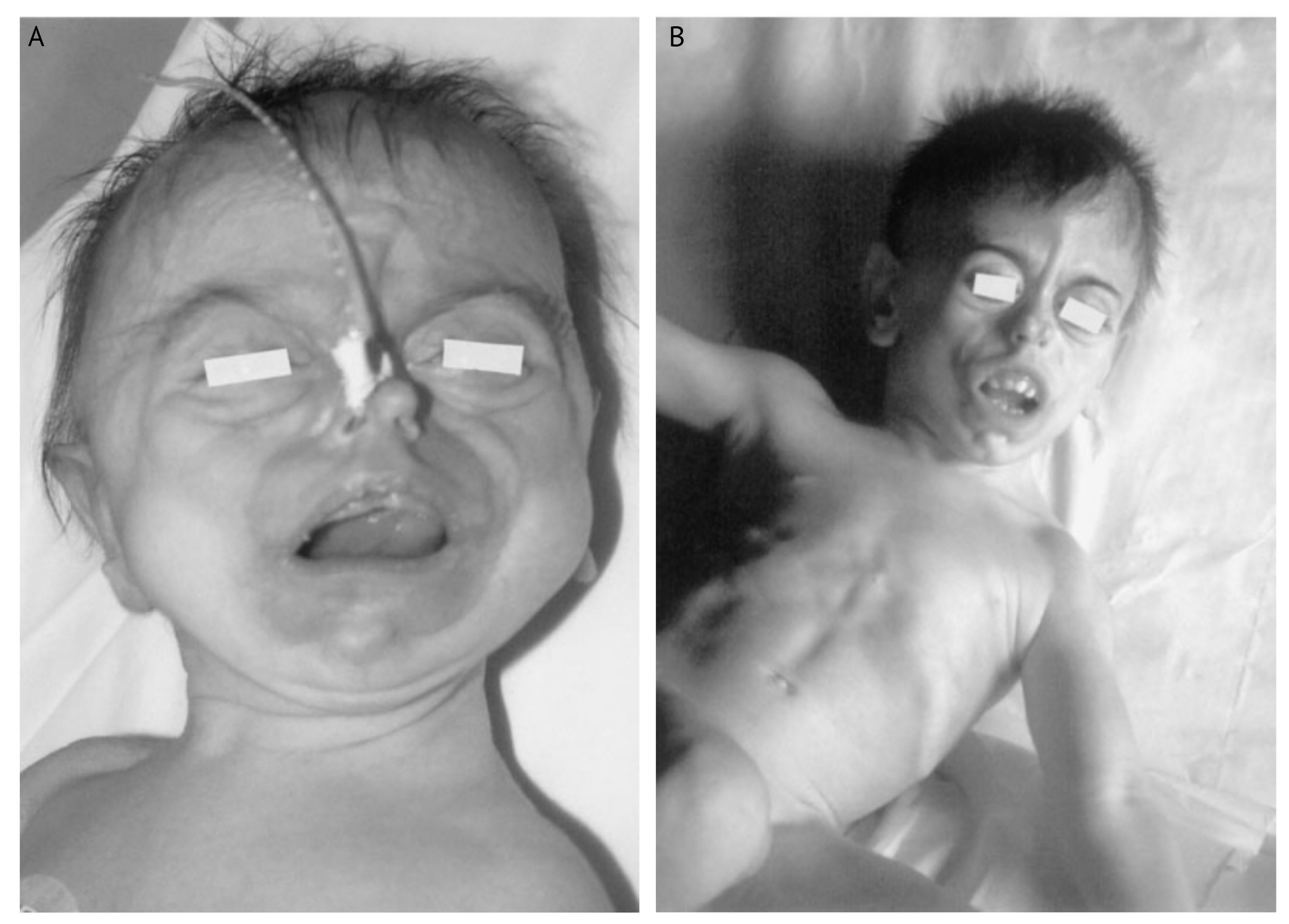
Синдром Кеппена-Любински — это крайне редкая каналопатия, вызываемая гетерозиготными мутациями в селективном фильтре изоформы GIRK2 [99]. В медицинской литературе описано всего три случая этого синдрома [100–102]. Их объединяют нормальные размеры тела при рождении с тяжелой задержкой роста и умственного развития впоследствии, липодистрофия с полным отсутствием подкожного жира на лице, специфические черты лица: узкая спинка носа и ноздри, натянутая верхняя губа, открытый рот, готическое небо, выступающие глаза в неглубоких глазницах. В более старшем возрасте тонус мышц конечностей повышен, сухожильные рефлексы усилены.
Все эти пациенты не связаны родством, но удивительно, что в двух случаях из трех возникли одинаковые делеции одной аминокислоты Thr152, а в третьем случае аминокислотная замена GIRK2Gly154Ser идентична той, которая вызывает фенотип weaver (англ. «ткач») у мышей, впервые описанный в 1964 году [103]. Для этих мышей характерна атаксия из-за дегенерации клеток-зерен в мозжечке. Считается, что клетки мозжечка не дифференцируются и деградируют вследствие постоянной деполяризации, вызванной увеличенной проводимостью для Na+ у мутантного GIRK2Gly156Ser.
Мутации Kir3.4: первичный гиперальдостеронизм
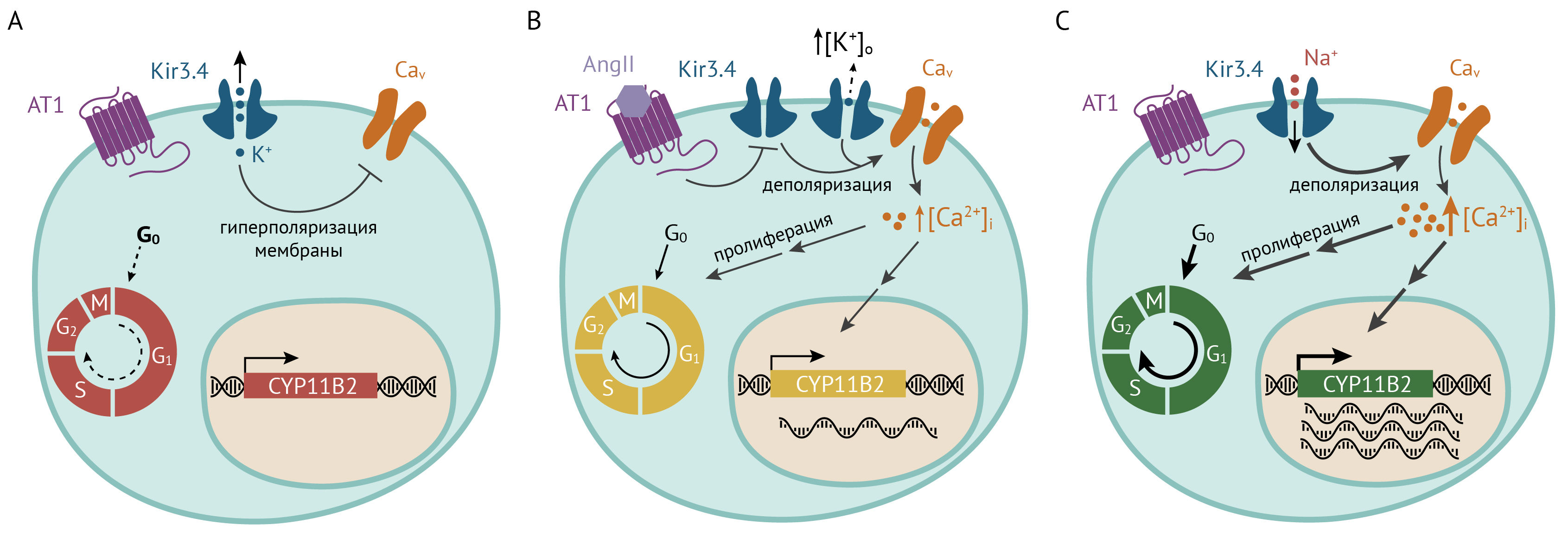
У пациентов с первичным гиперальдостеронизмом (синдромом Конна) были обнаружены мутации в гене KCNJ5, усиливающие проводимость канала для Na+. Такие мутации могут быть как генеративными — и тогда они вызывают раннюю гипертензию и гиперплазию коры надпочечников [104, 105], так и соматическими — в этом случае из мутантных клеток развивается производящая альдостерон аденома, и избыток альдостерона также ведет к гипернатриемии и гипертензии [106]. Механизм развития гиперальдостеронизма основан на том, что мутантный канал с нарушенным селективным фильтром пропускает Na+, мембрана деполяризуется, и мембранный потенциал достигает порогового для потенциалзависимых Ca2+-каналов значения. Активация Cav-каналов приводит к повышению [Ca2+]i и усилению экспрессии альдостеронсинтазы. А поскольку повышение внутриклеточной концентрации Ca2+ стимулирует пролиферацию клеток, в случае соматических мутаций развивается гиперплазия коры надпочечников или аденома.
Мутации Kir3.4: синдром удлиненного интервала QT (LQT13)
Мутации в гене KCNJ5 с утратой функции могут приводить к наследственному синдрому удлиненного интервала QT (OMIM: 613485) [107]. Утрата Kir3.4 в сердце затрудняет реполяризацию желудочков, что отражается на ЭКГ в виде удлинения QT. Однако распространенность среди пациентов с этим синдромом носителей мутаций в KCNJ5, вероятно, низка: до настоящего времени была обнаружена только две семьи с мутацией в Kir3.4 [107,108], а поиск среди 63 неродственных индивидов не дал результатов [109].
Источники:
- Morishige K.I. et al. Secretagogue-induced exocytosis recruits G protein-gated K+ channels to plasma membrane in endocrine cells // J. Biol. Chem. 1999. Vol. 274, № 12. P. 7969–7974.
- Oh U., Ho Y.K., Kim D. Modulation of the serotonin-activated K+ channel by G protein subunits and nucleotides in rat hippocampal neurons // J. Membr. Biol. 1995. Vol. 147, № 3. P. 241–253.
- Lacey M.G., Mercuri N.B., North R.A. On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurons // J. Physiol. 1988. Vol. 401, № 1. P. 437–453.
- Grudt T.J., Williams J.T. κ-Opioid receptors also increase potassium conductance. // Proc. Natl. Acad. Sci. 1993. Vol. 90, № 23. P. 11429–11432.
- North R.A. et al. μ- and δ- receptors belong to a family of receptors that are coupled to potassium channels. // Proc. Natl. Acad. Sci. 1987. Vol. 84, № 15. P. 5487–5491.
- Kubo Y. et al. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel // Nature. 1993. Vol. 364, № 6440. P. 802–806.
- Dascal N. et al. Atrial G protein-activated K+ channel: expression cloning and molecular properties. // Proc. Natl. Acad. Sci. 1993. Vol. 90, № 21. P. 10235–10239.
- Krapivinsky G. et al. Gβγ binds directly to the G protein-gated K+ channel, IKACh // J. Biol. Chem. 1995. Vol. 270, № 49. P. 29059–29062.
- Wischmeyer E. et al. Subunit interactions in the assembly of neuronal Kir3.0 Inwardly rectifying K+ channels // Mol. Cell. Neurosci. 1997. Vol. 9, № 3. P. 194–206.
- Spauschus A. et al. A G-protein-activated inwardly rectifying K+ channel (GIRK4) from human hippocampus associates with other GIRK channels. // J. Neurosci. 1996. Vol. 16, № 3. P. 930–938.
- Philipson L.H. et al. Functional expression of an epitope-tagged G protein-coupled K+ channel (GIRK1) // J. Biol. Chem. 1995. Vol. 270, № 24. P. 14604–14610.
- Ma D. et al. Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart // Neuron. 2002. Vol. 33, № 5. P. 715–729.
- Kennedy M.E., Nemec J., Clapham D.E. Localization and interaction of epitope-tagged GIRK1 and CIR inward rectifier K+ channel subunits // Neuropharmacology. 1996. Vol. 35, № 7. P. 831–839.
- Lesage F. et al. Cloning provides evidence for a family of inward rectifier and G-protein coupled K+ channels in the brain // FEBS Lett. 1994. Vol. 353, № 1. P. 37–42.
- Inanobe A. et al. Molecular cloning and characterization of a novel splicing variant of the Kir3.2 subunit predominantly expressed in mouse testis // J. Physiol. 1999. Vol. 521, № 1. P. 19–30.
- Wickman K., Pu W.T., Clapham D.E. Structural characterization of the mouse Girk genes // Gene. 2002. Vol. 284, № 1–2. P. 241–250.
- Wei J. et al. Characterization of Murine Girk2 Transcript Isoforms: Structure and Differential Expression // Genomics. 2002. Vol. 51, № 3. P. 379–390.
- Inanobe A. et al. Characterization of G-Protein-Gated K + Channels Composed of Kir3.2 Subunits in Dopaminergic Neurons of the Substantia Nigra // J. Neurosci. 1999. Vol. 19, № 3. P. 1006–1017.
- Karschin C. et al. IRK(1–3) and GIRK(1–4) Inwardly Rectifying K+ Channel mRNAs Are Differentially Expressed in the Adult Rat Brain // J. Neurosci. 1996. Vol. 76, № 11. P. 3559–3570.
- Liao Y.J., Jan Y.N., Jan L.Y. Heteromultimerization of G-Protein-Gated Inwardly Rectifying K+ Channel Proteins GIRK1 and GIRK2 and Their Altered Expression in weaver Brain // J. Neurosci. 1996. Vol. 16, № 22. P. 7137–7150.
- Kobayashi T. et al. Molecular Cloning of a Mouse G-Protein-Activated K+ Channel (mGIRK1) and Distinct Distributions of 3 GIRK (GIRK1, 2 and 3) mRNAs in Mouse Brain // Biochem. Biophys. Res. Commun. 1995. Vol. 208, № 3. P. 1166–1173.
- Chung H.J. et al. Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels // Proc. Natl. Acad. Sci. 2009. Vol. 106, № 2. P. 629–634.
- Jelaciс T.M., Sims S.M., Clapham D.E. Functional expression and characterization of G-protein-gated inwardly rectifying K+ channels containing GIRK3 // J. Membr. Biol. 1999. Vol. 169, № 2. P. 123–129.
- Schoots O. et al. Co-expression of Human Kir3 Subunits Can Yield Channels with Different Functional Properties // Cell. Signal. 1999. Vol. 11, № 12. P. 871–883.
- Lesage F. et al. Molecular Properties of Neuronal G-protein-activated Inwardly Rectifying K+ Channels // J. Biol. Chem. 1995. Vol. 270, № 48. P. 28660–28667.
- Lunn M.-L. et al. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction // Nat. Neurosci. 2007. Vol. 10, № 10. P. 1249–1259.
- Jelacic T.M. et al. Functional and biochemical evidence for G-protein-gated inwardly rectifying K+ (GIRK) channels composed of GIRK2 and GIRK3 // J. Biol. Chem. 2000. Vol. 275, № 46. P. 36211–36216.
- Krapivinsky G. et al. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins // Nature. 1995. Vol. 374, № 6518. P. 135–141.
- Tucker S.J., Pessia M., Adelman J.P. Muscarine-gated K+ channel: subunit stoichiometry and structural domains essential for G protein stimulation // Am. J. Physiol. Circ. Physiol. 1996. Vol. 271, № 1. P. H379–H385.
- Silverman S.K., Lester H.A., Dougherty D.A. Subunit stoichiometry of a heteromultimeric G protein-coupled inward-rectifier K+ channel // J. Biol. Chem. 1996. Vol. 271, № 48. P. 30524–30528.
- Corey S. et al. Number and Stoichiometry of Subunits in the Native Atrial G-protein-gated K+ Channel, IKACh // Biochemistry. 1998. Vol. 273, № 9. P. 5271–5278.
- Corey S., Clapham D.E. Identification of native atrial G-protein-regulated inwardly rectifying K+ (GIRK4) channel homomultimers // J. Biol. Chem. 1998. Vol. 273, № 42. P. 27499–27504.
- Leopoldt D., Harteneck C., Nürnberg B. G Proteins endogenously expressed in Sf 9 cells: Interactions with mammalian histamine receptors // Naunyn. Schmiedebergs. Arch. Pharmacol. 1997. Vol. 356, № 2. P. 216–224.
- Wang W., Whorton M.R., MacKinnon R. Quantitative analysis of mammalian GIRK2 channel regulation by G proteins, the signaling lipid PIP2 and Na+ in a reconstituted system // Elife. 2014. Vol. 3. P. e03671.
- Peleg S. et al. Gαi controls the gating of the G protein-activated K+ channel, GIRK // Neuron. 2002. Vol. 33, № 1. P. 87–99.
- Riven I., Iwanir S., Reuveny E. GIRK Channel Activation Involves a Local Rearrangement of a Preformed G Protein Channel Complex // Neuron. 2006. Vol. 51, № 5. P. 561–573.
- Digby G.J., Sethi P.R., Lambert N.A. Differential dissociation of G protein heterotrimers // J. Physiol. 2008. Vol. 586, № 14. P. 3325–3335.
- Touhara K.K., MacKinnon R. Molecular basis of signaling specificity between GIRK channels and GPCRs // Elife. 2018. Vol. 7. P. 8–10.
- Ho I.H.M., Murrell-Lagnado R.D. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels // J. Physiol. 1999. Vol. 520, № 3. P. 645–651.
- Whorton M.R., MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium // Cell. 2011. Vol. 147, № 1. P. 199–208.
- Wickman K. et al. Abnormal heart rate regulation in GIRK4 knockout mice // Neuron. 1998. Vol. 20, № 1. P. 103–114.
- Perry C.A. et al. Predisposition to late-onset obesity in GIRK4 knockout mice // Proc. Natl. Acad. Sci. 2008. Vol. 105, № 23. P. 8148–8153.
- Bettahi I. et al. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh // J. Biol. Chem. 2002. Vol. 277, № 50. P. 48282–48288.
- Kurachi Y., Nakajima T., Sugimoto T.N. On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins // Pflügers Arch. - Eur. J. Physiol. 1986. Vol. 407, № 3. P. 264–274.
- Luján R., Aguado C. Localization and Targeting of GIRK Channels in Mammalian Central Neurons // Int. Rev. Neurobiol. 2015. Vol. 123. P. 161–200.
- Signorini S. et al. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. // Proc. Natl. Acad. Sci. 1997. Vol. 94, № 3. P. 923–927.
- Lüscher C. et al. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons // Neuron. 1997. Vol. 19, № 3. P. 687–695.
- Torrecilla M. et al. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons // J. Neurosci. 2002. Vol. 22, № 11. P. 4328–4334.
- Chen S.C. et al. Developmental expression of the GIRK family of inward rectifying potassium channels: Implications for abnormalities in the weaver mutant mouse // Brain Res. 1997. Vol. 778, № 2. P. 251–264.
- Pepper C., Henderson G. Opiates and opioid peptides hyperpolarize locus coeruleus neurons in vitro // Science (80-. ). 1980. Vol. 209, № 4454. P. 394–395.
- Williams J.T., Egan T.M., North R.A. Enkephalin opens potassium channels on mammalian central neurones // Nature. 1982. Vol. 299, № 5878. P. 74–77.
- Williams J.T., North R.A. Opiate-receptor interactions on single locus coeruleus neurones // Mol. Pharmacol. 1984. Vol. 26, № 3. P. 489–497.
- Koyrakh L. et al. Molecular and Cellular Diversity of Neuronal G-Protein-Gated Potassium Channels // J. Neurosci. 2005. Vol. 25, № 49. P. 11468–11478.
- https://elementy.ru/novosti_na...
- Lalive A.L. et al. Firing Modes of Dopamine Neurons Drive Bidirectional GIRK Channel Plasticity // J. Neurosci. 2014. Vol. 34, № 15. P. 5107–5114.
- Di Chiara G., Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. // Proc. Natl. Acad. Sci. 1988. Vol. 85, № 14. P. 5274–5278.
- Moghaddam B., Bunney B.S. Differential effect of cocaine on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens: Comparison to amphetamine // Synapse. 1989. Vol. 4, № 2. P. 156–161.
- Bradberry C.W., Roth R.H. Cocaine increases extracellular dopamine in rat nucleus accumbens and ventral tegmental area as shown by in vivo microdialysis // Neurosci. Lett. 1989. Vol. 103, № 1. P. 97–102.
- Johnson S.W., North R.A. Opioids excite dopamine neurons by hyperpolarization of local interneurons. // J. Neurosci. 1992. Vol. 12, № 2. P. 483–488.
- Lüscher C., Ungless M.A. The Mechanistic Classification of Addictive Drugs // PLoS Med. 2006. Vol. 3, № 11. P. e437.
- Sulzer D. et al. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. // J. Neurosci. 1995. Vol. 15, № 5 Pt 2. P. 4102–4108.
- Chen R. et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter // Proc. Natl. Acad. Sci. 2006. Vol. 103, № 24. P. 9333–9338.
- Rocha B.A. Stimulant and reinforcing effects of cocaine in monoamine transporter knockout mice // Eur. J. Pharmacol. 2003. Vol. 479, № 1–3. P. 107–115.
- Arora D. et al. Altered neurotransmission in the mesolimbic reward system of Girk-/- mice // J. Neurochem. 2010. Vol. 114, № 5. P. 1487–1497.
- Rifkin R.A., Moss S.J., Slesinger P.A. G Protein-Gated Potassium Channels: A Link to Drug Addiction // Trends Pharmacol. Sci. 2017. Vol. 38, № 4. P. 378–392.
- Hearing M.C., Zink A.N., Wickman K. Cocaine-induced adaptations in metabotropic inhibitory signaling in the mesocorticolimbic system // Rev. Neurosci. 2012. Vol. 23, № 4. P. 325–351.
- Morikawa H., Morrisett R.A. Ethanol Action on Dopaminergic Neurons in the Ventral Tegmental Area. Interaction with Intrinsic Ion Channels and Neurotransmitter Inputs // International Review of Neurobiology. 2010. Vol. 91, № C. 235–288 p.
- Kobayashi T. et al. Ethanol opens G-protein-activated inwardly rectifying K+ channels // Nat. Neurosci. 1999. Vol. 2, № 12. P. 1091–1097.
- Morgan A.D. et al. Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice // Neuropsychopharmacology. 2003. Vol. 28, № 5. P. 932–938.
- Arora D. et al. Acute Cocaine Exposure Weakens GABAB Receptor-Dependent G-Protein-Gated Inwardly Rectifying K+ Signaling in Dopamine Neurons of the Ventral Tegmental Area // J. Neurosci. 2011. Vol. 31, № 34. P. 12251–12257.
- McCall N.M. et al. Selective Ablation of GIRK Channels in Dopamine Neurons Alters Behavioral Effects of Cocaine in Mice // Neuropsychopharmacology. 2017. Vol. 42, № 3. P. 707–715.
- Rossi M.A. et al. Operant Self-Stimulation of Dopamine Neurons in the Substantia Nigra // PLoS One. 2013. Vol. 8, № 6. P. 1–7.
- Ilango A. et al. Similar Roles of Substantia Nigra and Ventral Tegmental Dopamine Neurons in Reward and Aversion // J. Neurosci. 2014. Vol. 34, № 3. P. 817–822.
- Munoz M.B., Slesinger P.A. Sorting Nexin 27 Regulation of G Protein-Gated Inwardly Rectifying K+ Channels Attenuates In Vivo Cocaine Response // Neuron. 2014. Vol. 82, № 3. P. 659–669.
- Rifkin R.A. et al. GIRK currents in VTA dopamine neurons control the sensitivity of mice to cocaine-induced locomotor sensitization // Proc. Natl. Acad. Sci. 2018. Vol. 115, № 40. P. E9479–E9488.
- Munoz M.B. et al. A Role for the GIRK3 Subunit in Methamphetamine-Induced Attenuation of GABAB Receptor-Activated GIRK Currents in VTA Dopamine Neurons // J. Neurosci. 2016. Vol. 36, № 11. P. 3106–3114.
- Kajii Y. et al. A developmentally regulated and psychostimulant-inducible novel rat gene mrt1 encoding PDZ-PX proteins isolated in the neocortex // Mol. Psychiatry. 2003. Vol. 8, № 4. P. 434–444.
- Padgett C.L. et al. Methamphetamine-Evoked Depression of GABAB Receptor Signaling in GABA Neurons of the VTA // Neuron. 2012. Vol. 73, № 5. P. 978–989.
- Ungless M.A. et al. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons // Nature. 2001. Vol. 411, № 6837. P. 581–583.
- Meye F.J. et al. Cocaine-evoked negative symptoms require AMPA receptor trafficking in the lateral habenula // Nat. Neurosci. 2015. Vol. 18, № 3. P. 376–380.
- Lecca S. et al. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice // Nat. Med. 2016. Vol. 22, № 3. P. 254–261.
- Cruz H.G. et al. Bi-directional effects of GABAB receptor agonists on the mesolimbic dopamine system // Nat. Neurosci. 2004. Vol. 7, № 2. P. 153–159.
- Hearing M. et al. Repeated Cocaine Weakens GABAB-Girk Signaling in Layer 5/6 Pyramidal Neurons in the Prelimbic Cortex // Neuron. 2013. Vol. 80, № 1. P. 159–170.
- Sharpe A.L. et al. Methamphetamine Self-Administration in Mice Decreases GIRK Channel-Mediated Currents in Midbrain Dopamine Neurons // Int. J. Neuropsychopharmacol. 2015. Vol. 18, № 5. P. 1–10.
- Blednov Y. a et al. Potassium channels as targets for ethanol: studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice. 2001. Vol. 298, № 2. P. 521–530.
- Herman M.A. et al. GIRK3 gates activation of the mesolimbic dopaminergic pathway by ethanol // Proc. Natl. Acad. Sci. 2015. Vol. 112, № 22. P. 7091–7096.
- Kotecki L. et al. GIRK Channels Modulate Opioid-Induced Motor Activity in a Cell Type- and Subunit-Dependent Manner // J. Neurosci. 2015. Vol. 35, № 18. P. 7131–7142.
- Wickman K. et al. Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4 // J. Neurosci. 2000. Vol. 20, № 15. P. 5608–5615.
- Iizuka M. et al. Localization of a G-protein-coupled inwardly rectifying K+ channel, CIR, in the rat brain // Neuroscience. 1997. Vol. 77, № 1. P. 1–13.
- King B.M. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight // Physiol. Behav. 2006. Vol. 87, № 2. P. 221–244.
- Gao Q., Horvath T.L. Neuronal control of energy homeostasis // FEBS Lett. 2008. Vol. 582, № 1. P. 132–141.
- Yoshimoto Y. et al. Somatostatin induces hyperpolarization in pancreatic islet α cells by activating a G protein-gated K+ channel // FEBS Lett. 1999. Vol. 444, № 2–3. P. 265–269.
- Bond C.T. et al. Cloning and functional expression of the cDNA encoding an inwardly-rectifying potassium channel expressed in pancreatic β-cells and in the brain // FEBS Lett. 1995. Vol. 367, № 1. P. 61–66.
- Ferrer J. et al. Pancreatic Islet Cells Express a Family of Inwardly Rectifying K+ Channel Subunits Which Interact to Form G-protein-activated Channels // J. Biol. Chem. 1995. Vol. 270, № 44. P. 26086–26091.
- Abel K.B., Lehr S., Ullrich S. Adrenaline-, not somatostatin-induced hyperpolarization is accompanied by a sustained inhibition of insulin secretion in INS-1 cells. Activation of sulphonylurea K+ATP channels is not involved // Pflügers Arch. - Eur. J. Physiol. 1996. Vol. 432, № 1. P. 89–96.
- Iwanir S., Reuveny E. Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels // Pflügers Arch. - Eur. J. Physiol. 2008. Vol. 456, № 6. P. 1097–1108.
- Rorsman P. et al. Activation by adrenaline of a low-conductance G protein-dependent K+ channel in mouse pancreatic B cells // Nature. 1991. Vol. 349, № 6304. P. 77–79.
- Sieg A. et al. Epinephrine-induced hyperpolarization of islet cells without KATP channels // Am. J. Physiol. Metab. 2004. Vol. 286, № 3. P. E463–E471.
- Masotti A. et al. Keppen-Lubinsky Syndrome Is Caused by Mutations in the Inwardly Rectifying K+ Channel Encoded by KCNJ6 // Am. J. Hum. Genet. The American Society of Human Genetics, 2015. Vol. 96, № 2. P. 295–300.
- Basel-Vanagaite L., Shaffer L., Chitayat D. Keppen-Lubinsky syndrome: Expanding the phenotype // Am. J. Med. Genet. Part A. 2009. Vol. 149A, № 8. P. 1827–1829.
- De Brasi D. et al. New syndrome with generalized lipodystrophy and a distinctive facial appearance: Confirmation of Keppen-Lubinski syndrome? // Am. J. Med. Genet. Part A. 2003. Vol. 117A, № 2. P. 194–195.
- Gorlin, R.J., Cohen, M.M., Hennekam R.C.M. Syndromes of the Head and Neck. 4th ed. New York: Oxford University Press, 2001. 1283 p.
- Lane P.W. New mutation: weaver, wv // Mouse News Lett. 1964.
- Mulatero P. et al. KCNJ5 Mutations in European Families With Nonglucocorticoid Remediable Familial Hyperaldosteronism // Hypertension. 2011. Vol. 59, № 2. P. 235–240.
- Scholl U.I. et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5 // Proc. Natl. Acad. Sci. 2011. Vol. 109, № 7. P. 2533–2538.
- Choi M. et al. K+ Channel Mutations in Adrenal Aldosterone-Producing Adenomas and Hereditary Hypertension // Science (80-. ). 2011. Vol. 331, № 6018. P. 768–772.
- Yang Y. et al. Identification of a Kir3.4 Mutation in Congenital Long QT Syndrome // Am. J. Hum. Genet. 2010. Vol. 86, № 6. P. 872–880.
- Kokunai Y. et al. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1 // Neurology. 2014. Vol. 82, № 12. P. 1058–1064.
- Ackerman J.P. et al. Abstract 16277: Absence of Putative Long QT Syndrome (LQTS)-Causing Mutations in the KCNJ5-Encoded Kir3.4 Channel Among a Large Cohort of Unrelated Patients With LQT1-12 Genotype Negative/Phenotype Positive LQTS // Circulation. American Heart Association, 2011. Vol. 124, № suppl_21. P. A16277–A16277
