
Калиевые каналы внутреннего выпрямления: часть 4

В предыдущих частях мы рассматривали другие подсемейства калиевых каналов внутреннего выпрямления.
Часть 1 → https://medach.pro/post/2078
Часть 2 → https://medach.pro/post/2203
Часть 3 → https://medach.pro/post/2459
Kir6.x/SURx — АТФ-зависимые калиевые каналы
Для поддержания гомеостаза необходимы механизмы, отслеживающие энергетический статус клеток. Одним из сенсоров внутриклеточного уровня АТФ (ATP) являются АТФ-чувствительные калиевые каналы. Они закрываются в ответ на повышение внутриклеточной концентрации АТФ, повышая возбудимость мембраны. Таким образом KATP-каналы переводят сигнал о содержании энергетических эквивалентов в клетке с химического языка на электрический. Классическим примером модуляции электрической активности клеток KATP-каналами служит регуляция выделения инсулина бета-клетками островков Лангерганса в ответ на повышение концентрации глюкозы в плазме (ниже этот механизм рассмотрен детально).
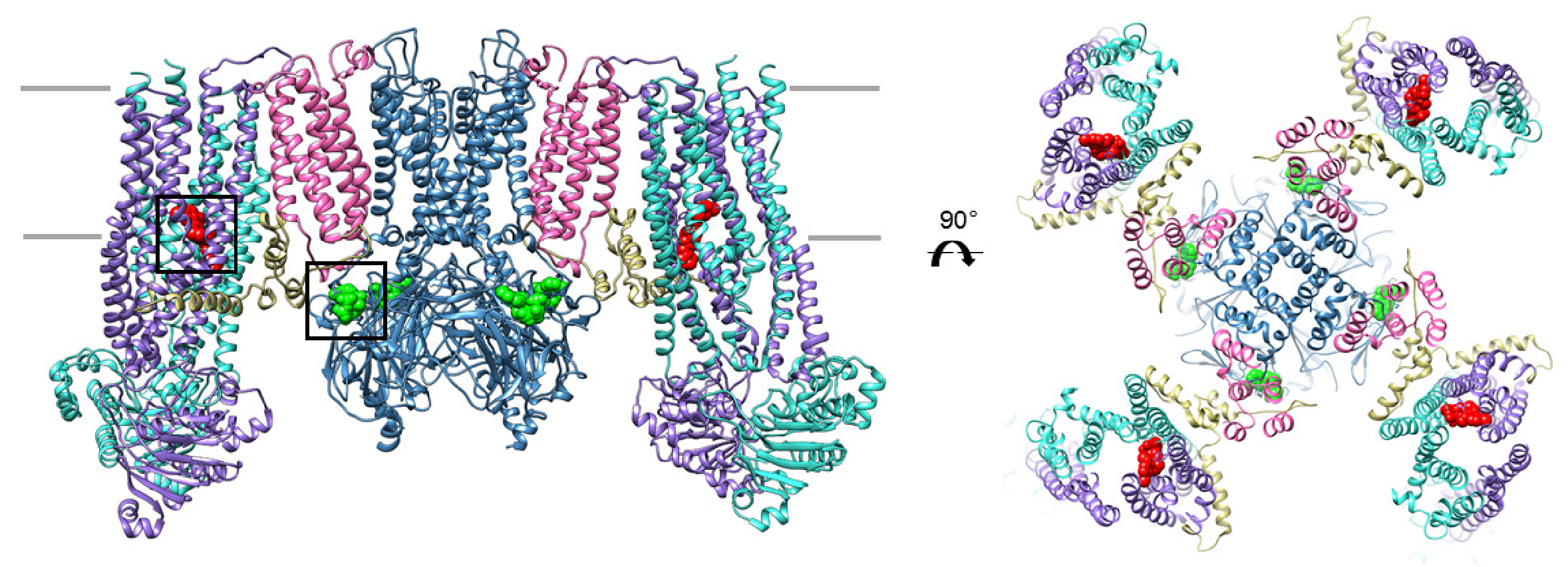
KATP-каналы — это гетерооктамеры из четырех субъединиц Kir6.x и четырех субъединиц SURx. SURx получили название «рецептор сульфонилмочевины» (sulphonylurea receptor), поскольку являются мишенью для противодиабетических препаратов класса производных сульфонилмочевины (ПСМ, таких как толбутамид, глибенкламид) и класса меглитинидов (натеглинид, репаглинид). Субъединицы Kir6.x образуют пору канала, а регуляторные субъединицы SUR располагаются по периферии. Общая структура этого канала приведена на рисунке 22.
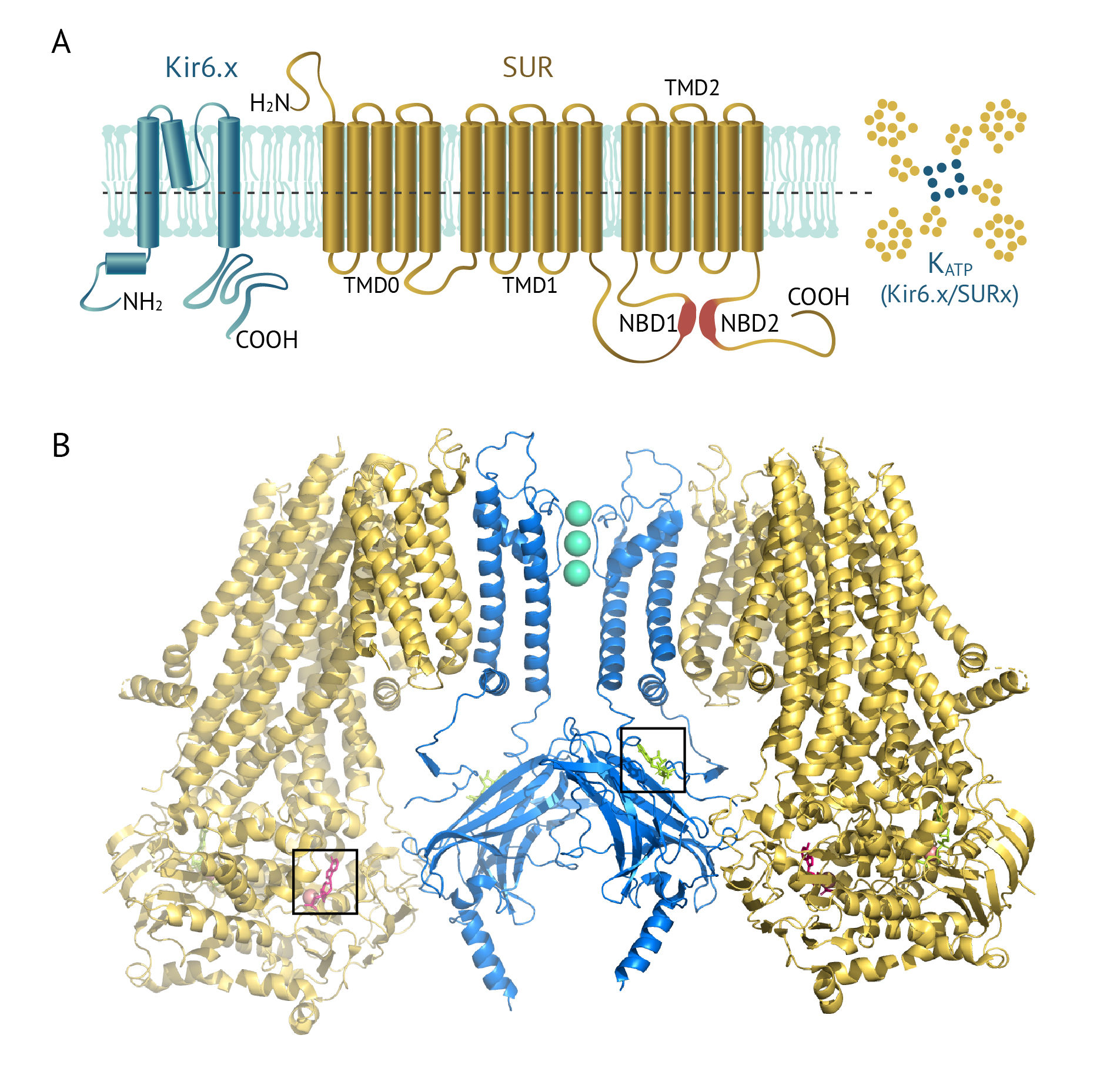
B. Структура двух пар SUR1-Kir6.2. SUR1 показаны желтым цветом, Kir6.2 — синим. В селективном фильтре канала расположены три иона K+, малиновым цветом показаны MgADP, салатовым — связанный с Kir6.2 АТФ [1].
В геноме человека есть два гена, кодирующие субъединицы Kir6: KCNJ8 кодирует Kir6.1, а KCNJ11 — Kir6.2 (прежнее название BIR — β-cell inward rectifier — канал внутреннего выпрямления в β-клетках); и два гена субъединиц SURx: ABCC8 (SUR1) и ABCC9 (SUR2). SURx относятся к семейству транспортеров АТФ-связывающей кассеты (ATP-binding cassette transporter); 17 пересекающих мембрану альфа-спиралей в их структуре организованы в три трансмембранных домена TMD0, TMD1 и TMD2.
SUR2 встречается в двух основных сплайс-вариантах SUR2A и SUR2B, которые различаются 42 C-концевыми остатками. Это небольшое отличие в структуре лежит в основе различий в функции и фармакологическом профиле Kir6.2/SUR2x каналов в различных тканях. Так, SUR2A экспрессируется в желудочках сердца, скелетных мышцах, яичниках, а также в нейронах головного мозга, языке и островках Лангерганса. SUR2B присутствует во многих тканях, например, в гладких мышцах и эндотелии сосудов, сократительной и проводящей системе сердца, эпителии легких, волосяных фолликулах, проксимальных канальцах почек, почечном эпителии, микроглии, астроцитах и в нейронах зубчатой извилины.
Регуляция KATP-каналов нуклеотидами и PIP2
KATP-каналы — сенсоры энергетического статуса клетки: они инактивируются в присутствии АТФ и активируются MgАДФ, причем сайты связывания АТФ и MgАДФ находятся на разных субъединицах (см. рис. 22).
Внутриклеточный АТФ блокирует KATP-каналы, связываясь с порообразующими субъединицами Kir6.x. Если удалить последние 26 аминокислот в Kir6.2, сигнал его локализации в ЭПР, можно получить независимые от SURx гомомерные каналы, которые блокируются АТФ (хотя их чувствительность к АТФ отличается) [2]. Существуют данные о том, что связывание АТФ дестабилизирует взаимодействие с PIP2 (про роль PIP2 в регуляции каналов Kir см. Часть 1) и таким образом стимулирует закрытие канала [3, 4].
За активацию KATP-каналов MgАДФ отвечают регуляторные субъединицы SURx. Между TMD1 и 2, а также на C-конце этих белков расположены нуклеотид-связывающие домены, которые содержат мотивы Walker A и B (названы так в честь открывшего их британского химика Джона Эрнеста Уокера). Мутации остатков лизина K719, K1384 (в SUR1) в мотивах Walker A и аспартата D854 в Walker B нарушают активацию KATP-каналов MgАДФ [5]. В этих же участках SURx связываются и лекарственные средства-активаторы калиевых каналов.
Активность KATP-каналов зависит от уровня PIP2, и эта связь может играть роль in vivo в сигналинге рецепторов, сопряженных с Gαq-белками: связывание лиганда с GPCR — рецепторами, сопряженными с G-белками (например, α1-адренорецептором, M1-холинорецепторами или рецепторами к эндотелину и ангиотензину II) ведет к активации PLC, которая расщепляет PIP2 [6].
Физиологическая роль KATP-каналов
Контроль уровня глюкозы с помощью KATP-каналов в β-клетках поджелудочной железы и в вентромедиальном гипоталамусе
В β-клетках KATP-каналы представлены гетеромерами Kir6.2/SUR1 [7]. При поступлении сахаров с пищей глюкоза из крови проникает в β-клетки поджелудочной железы через инсулиннезависимый транспортер GLUT2, где в результате ее катаболизма повышается соотношение [АТФ]/[АДФ]. АТФ связывается с KATP-каналами, в результате чего калиевый ток через них уменьшается, а мембрана деполяризуется. Когда деполяризация достигает порогового значения для потенциалзависимых кальциевых каналов L-типа (около −50 мВ), они открываются, Ca2+ поступает в клетку и запускает экзоцитоз везикул с инсулином [8].
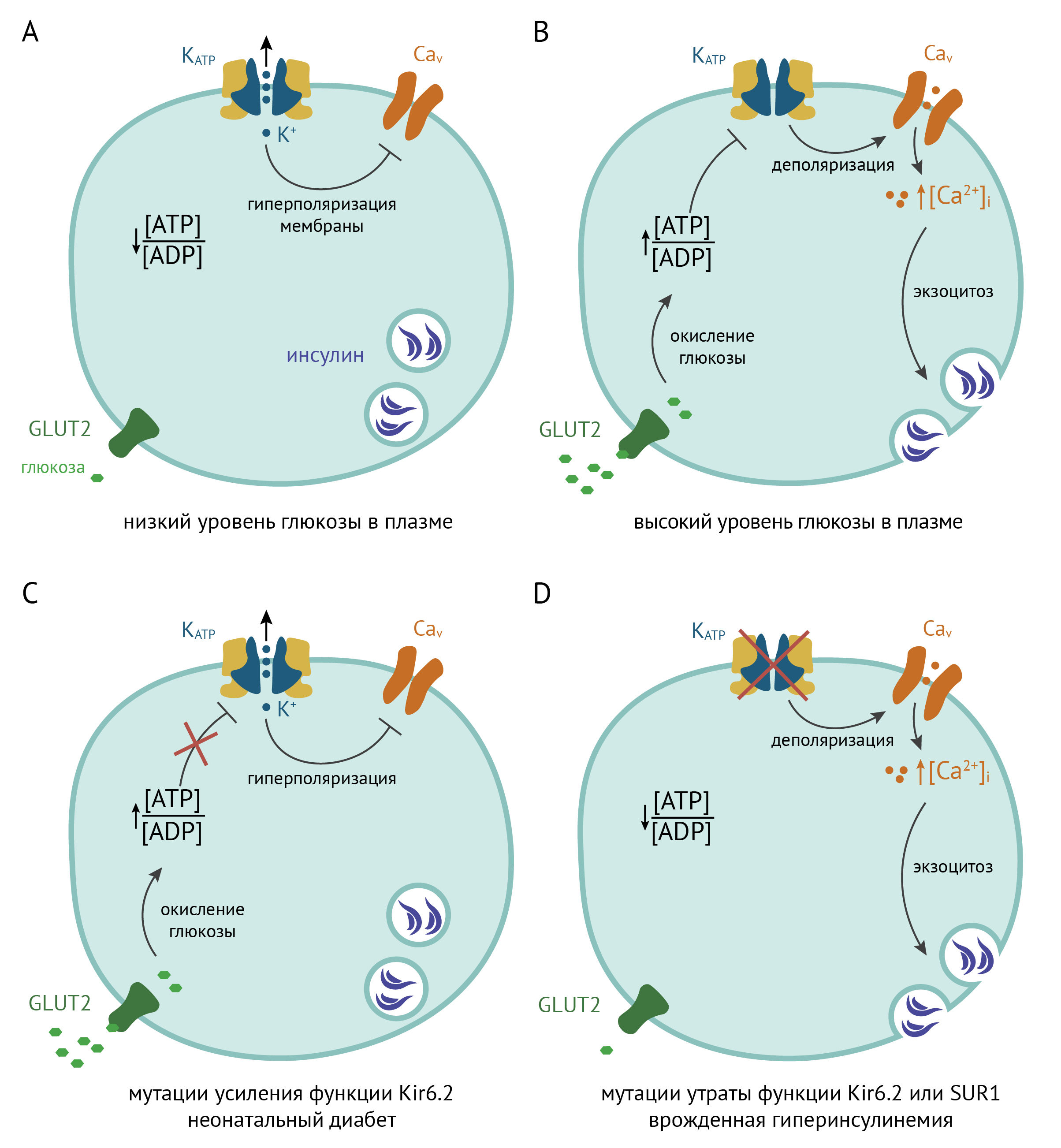
Рисунок 23 | KATP-каналы в клетках поджелудочной железы [9, с изменениями]
Исследования на трансгенных мышах, нокаутных по Kir6.2 или экспрессирующих доминантно-негативный вариант Kir6.2, свидетельствуют о роли KATP-каналов в дифференцировке клеток островков Лангерганса, в поддержании мембранного потенциала β-клеток и секреции инсулина в ответ на стимуляцию глюкозой. Новорожденные нокаутные мыши отличаются повышенным уровнем инсулина и гипогликемией, изолированные островки Лангерганса этих мышей не способны выделять инсулин в ответ на поступление глюкозы. В молодом возрасте концентрация глюкозы в крови у таких мышей нормальна и толерантность к глюкозе нарушена слабо. При внутрибрюшинном введении глюкозы или после приема пищи у нокаутных мышей наблюдается выделение небольшого количества инсулина, что, возможно, стимулируется инкретинами — глюкагоноподобным пептидом (GLP1) и гастроингибиторным пептидом (GIP), или другими механизмами. Однако с возрастом, если трансгенные мыши набирали вес, у них нарушалась толерантность к глюкозе и развивалась гипергликемия, чего не наблюдалось ни у нокаутных мышей с нормальной массой тела, ни у мышей дикого типа с ожирением [10]. Из исследований на мышах, нокаутных по Kir6.2, а также по рецепторам к GLP1 или GIP была предложена модель развития сахарного диабета II типа с первичным генетическим дефектом секреции инсулина, к которому затем подключается приобретенная инсулинорезистентность [10].
Секреция инсулина β-клетками зависит также от плотности KATP-каналов на мембране. При голодании плотность KATP-каналов на плазмалемме увеличивается через АМФК-зависимый сигнальный путь. Эта киназа активируется повышением соотношения [АМФ]/[АТФ] и стимулирует окисление жирных кислот в печени и скелетных мышцах и захват глюкозы миоцитами и тормозит синтез липидов и холестерола. Гормон жировой ткани лептин, помимо центральных эффектов на гипоталамус (снижает аппетит, действуя на латеральный гипоталамус, и стимулирует чувство насыщения, действуя на медиальный гипоталамус), стимулирует перемещение KATP-каналов на поверхность β-клеток, в этом сигнальном каскаде участвуют АМФК, PKA и CaMKKβ [11, 12].
В вентромедиальном ядре гипоталамуса (VMH) находятся нейроны, чувствительные к уровню глюкозы в мозге (так называемый центр насыщения). Как и β-клетки, нейроны VMH экспрессируют Kir6.2/SUR1 KATP-каналы, которые регулируют возбудимость этих нейронов в зависимости от метаболического статуса. В ответ на нейрогликопению эти нейроны через симпатическое звено автономной нервной системы запускают выделение глюкагона α-клетками поджелудочной железы. У мышей, нокаутных по Kir6.2 (Kcnj11−/−), секреция глюкагона в ответ на понижение концентрации глюкозы не нарушена, что было показано в экспериментах на изолированных островках Лангерганса, однако нейроны VMH не изменяют частоту генерации потенциалов действия в ответ на стимуляцию глюкозой [13].
Уровень глюкозы плазмы крови может регулироваться через активацию или торможение глюконеогенеза в печени, и в этом также участвуют KATP-каналы в гипоталамусе. Так, активация этих каналов (а также активация инсулинового рецептора) в нейронах аркуатного ядра ведет к ингибированию глюконеогенеза в печени и снижению концентрации глюкозы в плазме. Под действием инсулина активируется сигнальный каскад PI3K/Akt, который и открывает KATP-каналы, что ведет к гиперполяризации мембраны нейронов аркуатного ядра (схема). KATP-каналы есть как на проопиомеланокортиновых нейронах, так и на нейронах, экспрессирующих агути-родственный пептид [14], которые посылают проекции к ядрам блуждающего нерва в стволе мозга. У мышей, нокаутных по SUR1, стимуляция нейронов аркуатного ядра инсулином не приводила к подавлению глюконеогенеза, как и у мышей дикого типа с перерезанной печеночной ветвью блуждающего нерва [15]. Кажется удивительным, что KATP-каналы, стимулом к открытию которых обычно является недостаток энергии, в нейронах аркуатного ядра опосредуют влияние инсулина, связанное с подавлением высвобождения глюкозы, однако этот механизм был показан как на грызунах, так и на людях [16, 17]. Схожие KATP-зависимые эффекты наблюдались в ответ на ингибирование окисления жирных кислот, что свидетельствует о роли аркуатного ядра гипоталамуса в интеграции сигналов о метаболическом статусе [18, 19]. Этот механизм влияет не только на метаболизм гепатоцитов, но и на пищевое поведение. Нокаут гена PIP3 3-фосфатазы Pten в проопиомеланокортиновых нейронах аркуатного ядра вызывал стабильную активацию KATP-каналов в этих нейронах, и нокаутные мыши страдали от гиперфагии и ожирения [20]. Подробнее про центральную регуляцию потребления пищи можно прочитать здесь.
KATP-каналы в сердце
KATP-каналы присутствуют в клетках миокарда желудочков, предсердий и в проводящей системе сердца. В миокарде желудочков KATP-каналы состоят из Kir6.2/SUR2A субъединиц [21, 22][1]. Эти каналы имеют проводимость около 80 пСм [23, 24], блокируются глибенкламидом и открываются под действием активаторов калиевых каналов пинацидила и кромакалима, но практически нечувствительны к диазоксиду в отсутствие MgАДФ [25].
KATP-каналы предсердий несколько отличаются от желудочковых. Как и в желудочках, Kir6.2 является основной Kir6 субъединицей в предсердии; в кардиомиоцитах нокаутов по Kcnj11 KATP-ток отсутствует [26]. Однако, в отличие от желудочков, SUR представлен изоформой SUR1: у мышей, нокаутных по SUR1, ток через KATP-каналы отсутствует в кардиомиоцитах предсердий, но нормален в кардиомиоцитах желудочков [27]. Однако ключевая роль SUR1 в формировании KATP-каналов в предсердиях может быть характерна для грызунов, но не для человека, поскольку различия чувствительности к диазоксиду в атриальных и вентрикулярных кардиомиоцитах выражено слабо [28].
KATP-каналы в кардиомиоцитах в покое закрыты; они открываются в условиях гипоксии и ишемии, когда соотношение [АТФ]/[АДФ] падает, и сокращают длительность потенциала действия и эффективного рефрактерного периода [29]. Кроме того, KATP-каналы предсердий могут активироваться в гипотонической среде [30] или при растяжении мембраны [31]. Эта активация калиевого тока может сдерживать выделение предсердного натрийуретического пептида (ANP) в ответ на механическое растяжение: у мышей, нокаутных по Kir6.2, увеличение объема циркулирующей крови вызывает более интенсивное выделение ANP предсердиями, чем у мышей дикого типа [26]. Таким образом, KATP-каналы в предсердиях могут служить механизмом отрицательной обратной связи, ограничивая ответ на гиперволемию.
В проводящей системе сердца KATP-каналы могут иметь иной субъединичный состав, чем в желудочках и предсердиях: у мышей в клетках проводящей системы обнаружена экспрессия Kir6.1 помимо Kir6.2 [24], и варианты гена KCNJ8, ассоциированные с риском различных нарушений сердечного ритма [32–37]. В работе Bao и соавт. [24] также была обнаружена экспрессия изоформы SUR2B и отсутствие SUR1 в клетках проводящей системы. KATP-каналы участвуют в замедлении сердечного ритма в условиях гипоксии и могут запускать нарушения проведения возбуждения при ишемии [29].
KATP-каналы в кровеносных сосудах
KATP-каналы экспрессируются как в гладкомышечных клетках сосудов, так и в эндотелии.
В гладких мышцах сосудов KATP-каналы состоят преимущественно из Kir6.1/SUR2B [22, 38, 39] и также называются KNDP-каналами, т. к. для их активности необходимы нуклеозиддифосфаты в цитозоле. Эти каналы опосредуют вазодилатацию в ответ на аденозин [39], гипоксию [40] и физическую активность [41, 42], а также тормозят секрецию эндотелина [43].
У мышей, нокаутных по SUR2 [44] или Kir6.1 [38], повышено артериальное давление в покое и они спонтанно погибают от вазоспазма коронарных артерий, который сопровождается подъемом ST-сегмента. Это состояние может быть купировано антагонистом кальциевых каналов L-типа нифедипином. Симптомы у этих мышей очень сходны со стенокардией Принцметала (вариантной стенокардией) человека, однако мутации генов субъединиц KATP-каналов у пациентов с этим заболеванием до настоящего времени не найдены.
В общем, роль KATP-каналов в сердечно-сосудистой системе можно описать как кардиопротекторную и вазодилатирующую. Открытие KATP-каналов при ишемии приводит к укорочению ПД, уменьшению поступления кальция в цитозоль и отрицательному инотропному эффекту. Таким образом потребление АТФ снижается и сохраняются внутриклеточные запасы энергии, что противодействует ишемическому повреждению. Эта гипотеза экономии АТФ была высказана в самой первой публикации про сердечные KATP-каналы [45], и к настоящему времени каждый этап этого пути нашел экспериментальные подтверждения. Кардиопротекторная роль KATP-каналов связана с явлением прекондиционирования — повышенной устойчивости подвергшегося кратковременной ишемии сердца к последующим ишемическим атакам. Кроме того, прекондиционирование может быть вызвано адреналином, физической активностью и газообразными анестетиками. Антагонисты KATP-каналов блокируют кардиопротекторые эффекты прекондиционирования, а активаторы KATP-каналов имитируют прекондиционирование без гипоксии [29]. KATP-каналы могут способствовать сохранению функций митохондрий при ишемии: либо напрямую через митохондриальные KATP-каналы[2] (O’Rourke, 2000), либо опосредованно, через снижение поступления Ca2+ в цитозоль (Storey, 2013). Предполагается, что поступление K+ в митохондрии при ишемии и работа K+/H+-антипортера позволяет увеличить объем матрикса митохондрий и стимулирует синтез АТФ [46]. Митохондрии, изолированные из подвергшегося ишемии сердца, синтезируют АТФ более интенсивно [47]. В защите миокарда от ишемии участвуют не только KATP-каналы сердца, но и каналы других локализаций: каналы гладких мышц и эндотелия сосудов вызывают вазодилатацию, KATP-каналы нервных окончаний симпатической нервной системы тормозят выброс норадреналина, что удлиняет диастолу и снижает потребность сердца в кислороде [48].
Мутации и полиморфизмы в KATP-каналах человека
Мутации и полиморфизмы в генах порообразующих (KCNJ8, 11) и регуляторных (ABCC8, 9) субъединиц KATP-каналов вызывают различные метаболические нарушения, сердечно-сосудистые заболевания и аномалии развития.
Семейная гиперинсулинемическая гипогликемия
Мутации, нарушающие работу Kir6.2/SUR1, вызывают семейную гиперинсулинемическую гипогликемию (OMIM: 601820 и 256450). Дефектные KATP-каналы способствуют постоянной деполяризации мембраны β-клеток и секреции инсулина даже при низком уровне глюкозы в плазме (см. рис. 23). Симптомы проявляются у новорожденных и включают в себя высокую массу при рождении для данного срока, судороги и кому [49]. Иногда гиперинсулинемию удается скорректировать диазоксидом или соматостатином, однако во многих случаях приходится прибегать к удалению части поджелудочной железы. В различных популяциях генетические основы этого состояния различаются. Так, в Саудовской Аравии значительная доля случаев вызвана одной из двух мутаций в SUR1 [50], среди евреев ашкенази 88 % случаев объясняется присутствием одного из двух мутантных аллелей ABCC8 [51], в Финляндии большая часть случаев этого заболевания кластеризуются в одном регионе и объясняются одной мутацией SUR1V147D [52], однако в целом разнообразие мутаций велико [53–55]. Характер наследования обычно аутосомно-рецессивный, но бывают и доминантные мутации. Мутации, которые связаны с аутосомно-доминантным наследованием, обычно вызывают более легкую форму заболевания и проявляются позже, а часть носителей асимптоматичны [56].
Неонатальный сахарный диабет и предрасположенность к диабету II типа
Мутации приобретения функции в генах субъединиц Kir6.2 и SUR1 могут приводить к неонатальному сахарному диабету. Диабет новорожденных подразделяется на перманентный (OMIM: 606176) и транзиторный (OMIM: 610582 и 610374). В первом случае гипергликемия наблюдается с первых месяцев жизни и требует непрерывной терапии, а во втором уровень глюкозы в плазме приходит в норму с возможным возобновлением симптомов в более взрослом возрасте. Перманентный неонатальный сахарный диабет может проявляться или только в форме метаболического нарушения, или дополнительно включать в себя задержку развития и эпилепсию, тогда говорят о DEND-синдроме (developmental delay, epilepsy, and neonatal diabetes). В общем случае диабет новорожденных вызывают мутации в KCNJ11 и ABCC8, снижающие чувствительность KATP-каналов к ингибированию АТФ и повышающие вероятность открытия канала (Po). Такие каналы не закрываются в ответ на повышение [АТФ] и постоянно гиперполяризуют мембрану клетки. Интересно, что каналы с подобными мутациями как в KCNJ11, так и в ABCC8 сохраняют чувствительность к производным сульфонилмочевины, что позволяет использовать эти препараты вместо инсулина [57, 58, 67, 59–66].
Полиморфизм в гене KCNJ11, ведущий к аминокислотной замене Glu23Lys, часто присущ пациентам с диабетом II типа (OMIM: 125853) [68], и эти результаты были подтверждены на различных популяциях [69–71]. При замене глутамата на положительно заряженный лизин увеличивается вероятность открытия канала и уменьшается его чувствительность к АТФ [72], в результате чего снижается толерантность к глюкозе [73]. Этот вариант зачастую встречается вместе с полиморфизмом в кодоне 1369 гена ABCC8, заменой Ser1369Ala, поскольку гены KCNJ11 и ABCC8 расположены близко друг к другу [74]. В работе [75] авторы вызывали экспрессию редко встречающейся комбинации Lys23/Ser1369 и Glu23/Ala1369 и выяснили, что уменьшение чувствительности к АТФ происходит вследствие полиморфизма S1369A.
Синдромы с J-волной
Мутации усиления функции в KCNJ8 могут быть фактором развития синдромов с J-волной, к которым относятся синдром Бругада и синдром ранней реполяризации желудочков. В настоящее время описан один вариант Kir6.1Ser422Leu, связанный с синдромами с J-волной [32, 35, 36] и с повышенным риском развития фибрилляции предсердий [32, 34], однако причинно-следственная связь между этой мутацией и развитием синдромов с J-волной не ясна [78, 79]. Этот вариант чаще встречается в популяции ашкенази, среди них даже был описан гомозиготный по этому варианту индивид [37]. Из этого исследования может следовать, что вариант Kir6.1Ser422Leu — это безопасный полиморфизм, или что риск синдромов с J-волной повышен в популяции ашкенази, или что патогенетичность этого варианта зависит от генетического бэкграунда, который различается в разных европейских популяциях.
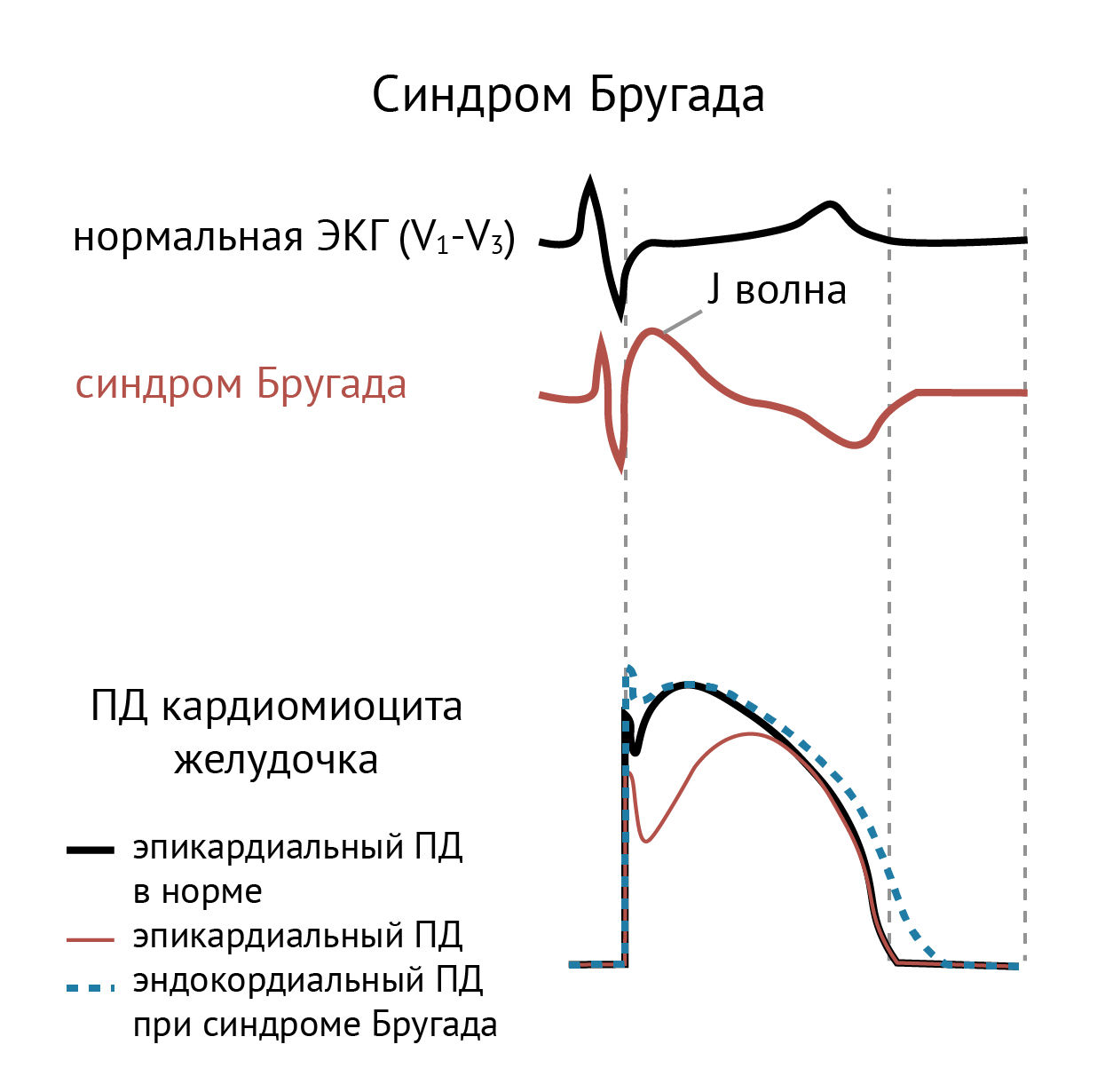
Рисунок 24 | Синдромы с J-волной на ЭКГ (отведения V1–V3) и особенности потенциала действия при синдроме Бругада [80]
Синдром Канту
Синдром Канту (OMIM: 239850) — это редкое генетическое нарушение, среди симптомов которого — врожденный гипертрихоз, макросомия при рождении, остеохондроплазия, кардиомегалия, открытый артериальный проток, расширенные и извилистые сосуды, специфические черты лица и др. (см. рис. 25) [81]. Этот синдром вызывают мутации в гене ABCC9, наследуемые по аутосомно-доминатному типу [82, 83], и у двоих пациентов без мутаций в ABCC9 были обнаружены мутации усиления функции в KCNJ8 (присутствуют в основном в гладких мышцах сосудов, но не в сердце), снижающие чувствительность канала к АТФ [79, 84]. Интересно, что сердечно-сосудистые проявления синдрома Канту возникают вследствие компенсаторных механизмов, и длительное применение глибенкламида подавляло гипертрофию сердца в модели синдрома Канту на мышах с мутацией SUR2Ala478Val и частично подавляло ее в модели с мутацией Kir6.1Val65Met с пониженной чувствительностью к глибенкламиду. Уровень глюкозы в плазме крови временно упал в начале применения глибенкламида, но вскоре вернулся в норму [85].
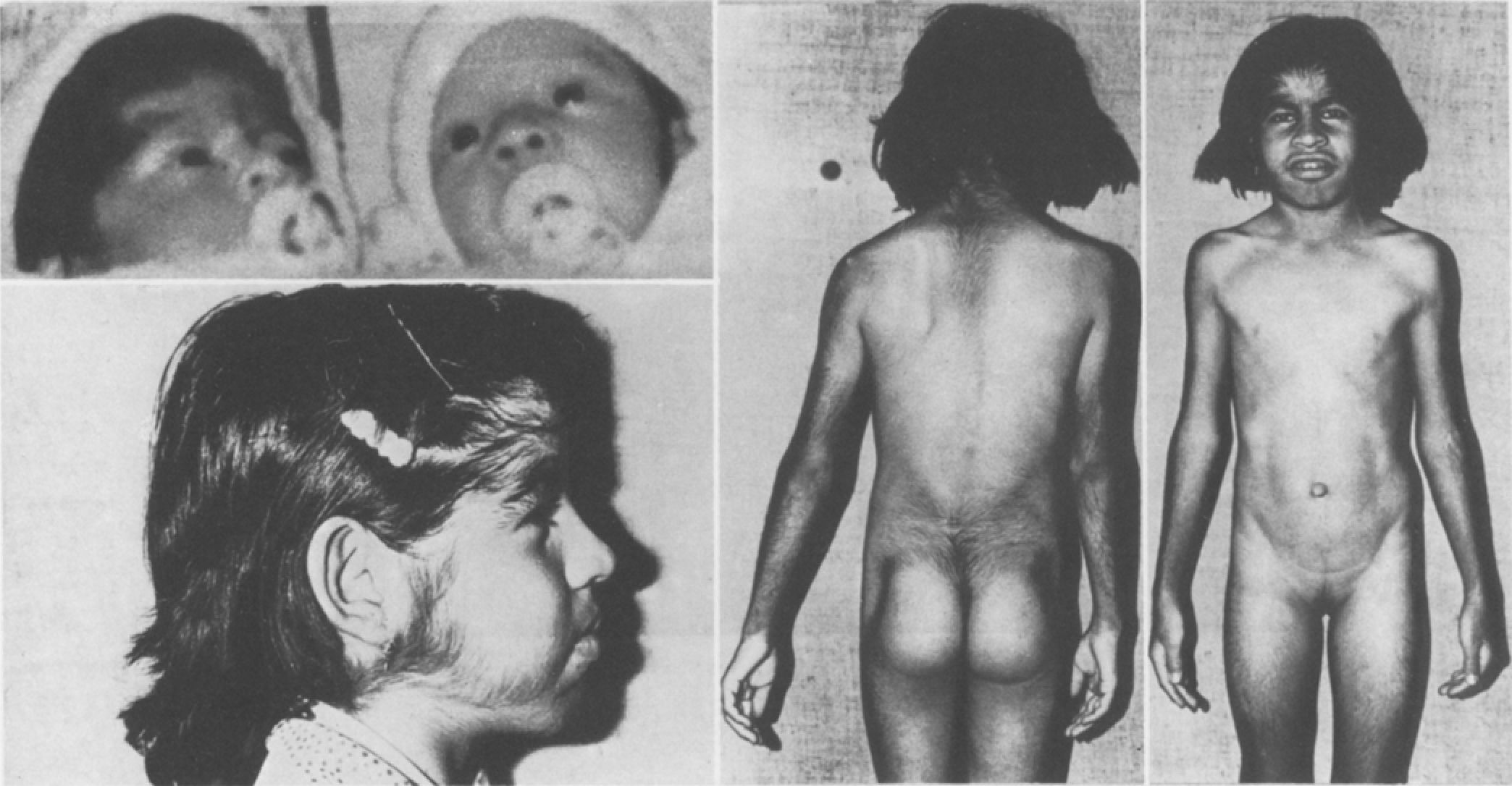
Рисунок 25 | Типичная внешность пациента с синдромом Канту при рождении и в возрасте 10 лет. На верхней левой панели — вместе со здоровой сестрой-близнецом [81]
Интересно, что часть симптомов синдрома Канту может возникать при использовании активатора KATP-каналов миноксидила: так, миноксидил стимулирует рост волос и топически применяется для контроля андрогенной алопеции [86]; описан случай гипертрихоза и аномалий развития у новорожденного после приема миноксидила при беременности [87], а в высоких дозировках миноксидил может вызывать псевдоакромегалию [88]. Предполагалось, что миноксидил улучшает кровообращение волосяных фолликулов, однако точный механизм влияния миноксидила на рост волос и мягких тканей неизвестен.
Дилатационная кардиомиопатия
У двух пациентов с дилатационной кардиомиопатией были обнаружены гетерозиготные варианты в экзоне 38 гена ABCC9, который кодирует C-конец изоформы SUR2A (OMIM: 608569) [89]. Миссенс-мутация A1513T и мутация сдвига рамки считывания Fs1524 снижают каталитическую активность второго нуклеотид-связывающего домена и нарушают регуляцию воротного механизма канала нуклеотидами.
Синдром AIMS
В 2019 году была обнаружена мутация сплайс-сайта в гене ABCC9 (c.1320 +1G> A), которая в гомозиготном состоянии вызывала нарушение интеллектуального развития и миопатию. Этот синдром получил аббревиатуру AIMS (ABCC9-related Intellectual disability Myopathy Syndrome). Синдром был описан на основании шести пациентов из двух семей в Северной Норвегии. В гетерозиготном состоянии этот генетический вариант не был найден в Азии и в Африке, но встречается в европейских популяциях, и его частота достигает 0,07 % среди финнов. На молекулярном уровне этот вариант ведет к делеции экзона 8 в SUR2 и к полному отсутствию функциональных SUR2-содержащих KATP-каналов. Среди симптомов AIMS отмечается повышенная утомляемость, гиперинтенсивность белого вещества на МРТ, тревожность и систолическая дисфункция у двух старших пациентов. Мыши, нокаутные по гену Abcc9, также страдали от утомляемости и увеличения левого желудочка. У трех из шести пациентов был гипотелоризм, что согласуется с уменьшенным расстоянием между глазами у нокаутных Danio rerio, однако для оценки влияния экспрессии SUR2 на морфогенез черт лица необходимо обследовать больше пациентов [90].
Фармакология
Классические блокаторы потенциал-зависимых калиевых каналов — производные тетраэтиламмония (TEA) и 4-аминопиридина (4-AP) — слабо влияют на каналы Kir [91, 92]. Ионы Cs+ и Ba2+ в микромолярных концентрациях блокируют каналы Kir, причем при аппликации с внешней стороны мембраны их действие сильнее, если мембрана гиперполяризована, и тем слабее, чем выше внеклеточная концентрация K+ [91, 93]. Хотя природных селективных высокоаффинных блокаторов Kir-каналов не найдено, некоторые вещества могут блокировать ток через Kir-каналы.
Тертиапин — токсин, изолированный из пчелиного яда, — блокирует KG и Kir1.1. [94], а его мутантная форма с двумя заменами His12Leu, Met13Gln является сравнительно специфичным блокатором Kir1.1 [95].
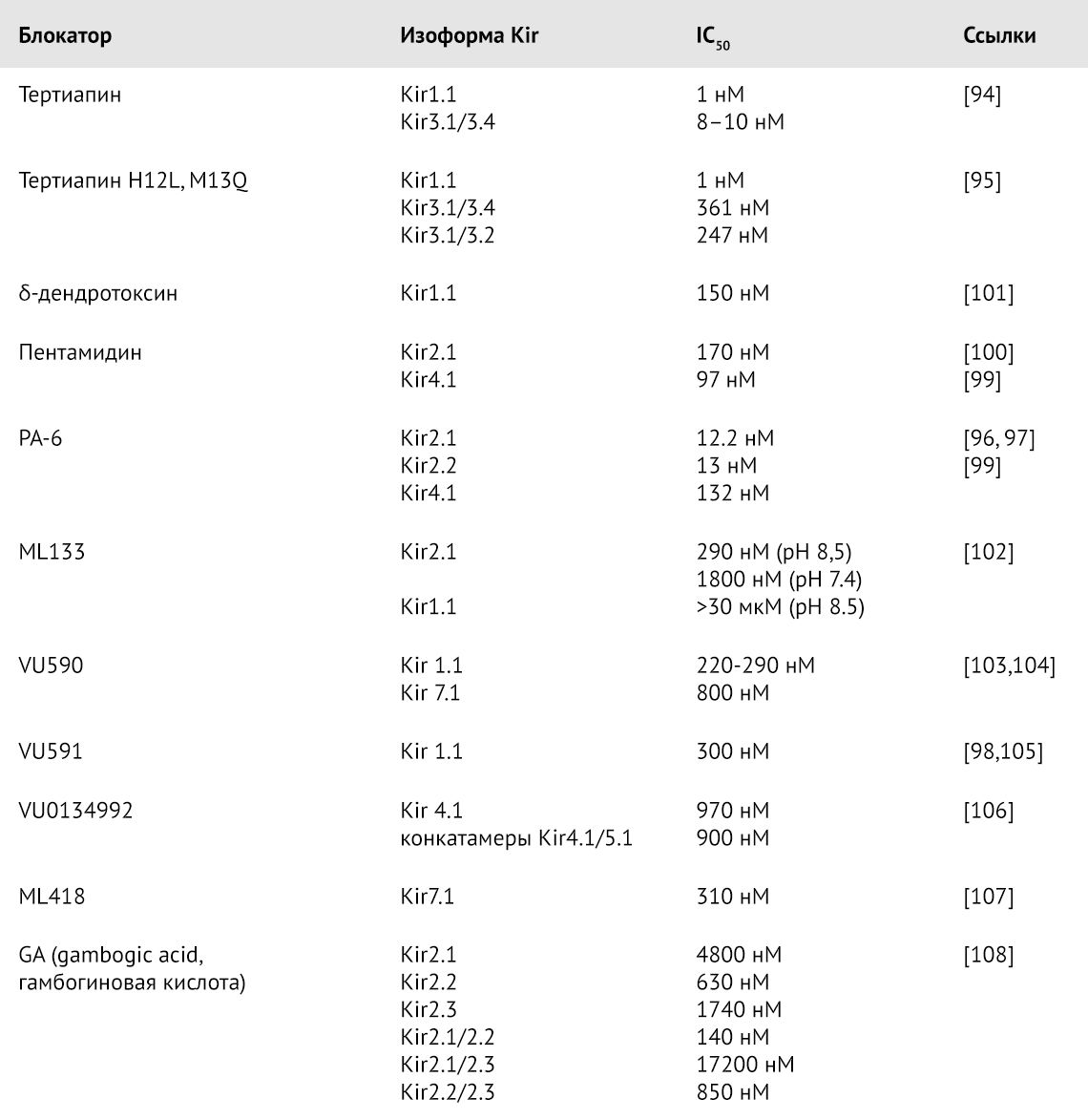
До недавнего времени селективных блокаторов каналов семейства Kir, действующих на порообразующие субъединицы, не существовало, однако в последние годы были найдены блокаторы с субмикромолярной концентрацией полумаксимального ингибирования (IC50) (таблица 1). Противопротозойный препарат пентамидин блокирует различные калиевые каналы, в том числе Kir2.x, что, вероятно, может объяснять удлинение интервала QT и другие аритмии при применении этого препарата. На его основе был получен его аналог PA-6, обладающий большей аффинностью к Kir2.x [96, 97]. Для некоторых ингибиторов известны сайты связывания с каналами Kir, например, VU591 блокирует пору Kir1.1 в области Val168 и Asn171 [98]. Для блокирующего действия пентамидина нужны остатки Thr127, Thr128 и Glu158 в Kir4.1 [99] и Glu224, Asp259 и Glu299 в Kir2.1 [100].
Таблица 1 | Блокаторы калиевых каналов
Фармакологические агенты, влияющие на KATP-каналы, действуют через их бета-субъединицы SURx. Производные сульфонилмочевины (ПСМ) и меглитиниды блокируют KATP-каналы в разных сайтах связывания (подробнее про препараты можно почитать тут). Эффект ПСМ на KATP-каналы зависит от внутриклеточной концентрации MgАДФ, а также от изоформы SURx: толбутамид блокирует SUR1-содержащие каналы β-клеток в 10 раз сильнее в присутствии MgАДФ [5, 109], а SUR2-содержащие каналы не отвечают на 300 мкМ глибенкламида при внутриклеточной концентрации MgАДФ 100 мкМ [110]. Новые исследования структуры KATP-каналов могут объяснить механизм блокировки канала и зависимость ее эффективности от концентрации внутриклеточного MgАДФ. В 2017 году методом криоэлектронной микроскопии были изучены несколько структур канала Kir6.2/SUR1 в присутствии глибенкламида [111–113]. В этих структурах два гомологичных нуклеотид-связывающих домена SUR1 повернуты приблизительно на 15° относительно оси симметрии и не могут димеризоваться. Авторы этих работ предполагают, что либо глибенкламид стабилизирует эту «повернутую» конформацию и препятствует димеризации NBD-доменов, либо такая конформация присуща SUR1 в норме, а глибенкламид не дает конформационным изменениям при гидролизе передаться на поровый домен Kir6.2.
KATP-каналы также служат мишенью для активаторов калиевых каналов (K-channel openers), таких как миноксидил, кромакалим, диазоксид, никорандил и пинацидил [114]. Эти вещества также действуют на SURx-субъединицы и их эффекты зависят от изоформы SUR. Активаторы калиевых каналов различаются механизмами действия. Некоторые из них, такие как пинацидил, кромакалим и их производные, уменьшают чувствительность KATP-каналов к АТФ и стимулируют открытие каналов при фиксированной концентрации АТФ в цитозоле [29]. Пинацидил действует на SUR2 и не влияет на SUR1-содержащие KATP-каналы. Для действия активаторов калиевых каналов другой группы, диазоксида и никорандила, необходим внутриклеточный АДФ. Зависимость чувствительности KATP-каналов к активаторам и блокаторам от содержания внутриклеточных нуклеотидов необходимо учитывать в анализе функции KATP-каналов при различных метаболических состояниях.
Поскольку экспрессия различных вариантов SUR тканеспецифична, эффекты активаторов калиевых каналов несколько различаются. Так, все активаторы калиевых каналов вызывают вазодилатацию, действуя на гладкие мышцы сосудов, и поэтому используются в терапии артериальной гипертензии. Благодаря действию на SUR1 диазоксид может тормозить секрецию инсулина и используется при врожденном гиперинсулинизме или инсулиноме и противопоказан при сахарном диабете II типа. Миноксидил в настоящее время не используют как антигипертензивный препарат, но применяют местно для коррекции андрогенной алопеции (см. раздел синдром Канту).
Таблица 2 | Действие активаторов калиевых каналов на KATP-каналы в различных типах клеток [114]

Кальциевый сенсибилизатор левосимендан, используемый в терапии сердечной недостаточности, помимо тропонина C действует и на KATP-каналы. Левосимендан открывает KATP-каналы в присутствии АТФ с EC50 ~4 мкМ. Он действует на каналы в кардиомиоцитах предсердий [115] и желудочков, что ведет к укорочению ПД [116], а также в гладких мышцах сосудов [117], вызывая вазодилатацию.
[1] Есть данные об экспрессии Kir6.1 и SUR1 в кардиомиоцитах желудочков, которые могут участвовать в формировании атипичного KATP-канала с проводимостью 21 пСм, чувствительного к активации диазоксидом [118].
[2] В 2019 году группа ученых под руководством Диего Де Стефани обнаружила собственно митохондриальные KATP-каналы — mitoKATP, которые образованы субъединицами CCDC51 — MITOK и ABCB8 — MITOSUR [119].
Библиография
- Lee K.P.K., Chen J., MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP // Elife. 2017. Vol. 6. P. 1–23.
- Tucker S.J. et al. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor // Nature. 2003. Vol. 387, № 6629. P. 179–183.
- John S.A., Weiss J.N., Ribalet B. ATP sensitivity of ATP-sensitive K+ channels: Role of the γ phosphate group of ATP and the R50 residue of mouse Kir6.2 // J. Physiol. 2005. Vol. 568, № 3. P. 931–940.
- Shyng S.L., Nichols C.G. Membrane phospholipid control of nucleotide sensitivity of KATP channels // Science (80-. ). 1998. Vol. 282, № 5391. P. 1138–1141.
- Gribble F.M., Tucker S.J., Ashcroft F.M. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide // EMBO J. 1997. Vol. 16, № 6. P. 1145–1152.
- Haruna T. et al. alpha1-Adrenoceptor-Mediated Breakdown of Phosphatidylinositol 4,5-Bisphosphate Inhibits Pinacidil-Activated ATP-Sensitive K+ Currents in Rat Ventricular Myocytes // Circ. Res. 2002. Vol. 91, № 3. P. 232–239.
- Aguilar-Bryan L. et al. Toward Understanding the Assembly and Structure of KATP Channels // Physiol. Rev. 1998. Vol. 78, № 1. P. 227–245.
- Rorsman P., Ashcroft F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men // Physiol. Rev. 2018. Vol. 98, № 1. P. 117–214.
- Ashcroft F.M. ATP-sensitive potassium channelopathies: Focus on insulin secretion // J. Clin. Invest. 2005. Vol. 115, № 8. P. 2047–2058.
- Seino S. et al. Diverse roles of K(ATP) channels learned from Kir6.2 genetically engineered mice // Diabetes. 2000. Vol. 49, № 3. P. 311–318.
- Chen P.-C., Kryukova Y.N., Shyng S.-L. Leptin Regulates KATP Channel Trafficking in Pancreatic β-Cells by a Signaling Mechanism Involving AMP-activated Protein Kinase (AMPK) and cAMP-dependent Protein Kinase (PKA) // J. Biol. Chem. 2013. Vol. 288, № 47. P. 34098–34109.
- Park S.-H. et al. Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic β-cells // Proc. Natl. Acad. Sci. 2013. Vol. 110, № 31. P. 12673–12678.
- Miki T. et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis // Nat. Neurosci. 2001. Vol. 4, № 5. P. 507–512.
- Könner A.C. et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production // Cell Metab. 2007. Vol. 5, № 6. P. 438–449.
- Pocai A. et al. Hypothalamic KATP channels control hepatic glucose production // Nature. 2005. Vol. 434, № 7036. P. 1026–1031.
- Carey M. et al. Central KATP channels modulate glucose effectiveness in humans and rodents // Diabetes. 2020. Vol. 69, № 6. P. 1140–1148.
- Kishore P. et al. Activation of KATP channels suppresses glucose production in humans // J. Clin. Invest. 2012. Vol. 121, № 12. P. 4916–4920.
- Pocai A. et al. A brain-liver circuit regulates glucose homeostasis // Cell Metab. 2005. Vol. 1, № 1. P. 53–61.
- Ruud J., Steculorum S.M., Bruning J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism // Nat. Commun. 2017. Vol. 8, № May. P. 1–12.
- Plum L. et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity // J. Clin. Invest. 2006. Vol. 116, № 7. P. 1886–1901.
- Stoller D.A. et al. Cardiomyocyte sulfonylurea receptor 2-K ATP channel mediates cardioprotection and ST segment elevation // Am. J. Physiol. Circ. Physiol. 2018. Vol. 1, № 34. P. 1100–1108.
- Suzuki M. et al. Functional Roles of Cardiac and Vascular ATP-Sensitive Potassium Channels Clarified by Kir6.2-Knockout Mice // Circ. Res. 2001. Vol. 88. P. 570–577.
- Babenko A.P. et al. Reconstituted Human Cardiac KATP Channels // Circ. Res. 2012. Vol. 83, № 11. P. 1132–1143.
- Bao L. et al. Unique properties of the ATP-sensitive K+ channel in the mouse ventricular cardiac conduction system // Circ. Arrhythmia Electrophysiol. 2011. Vol. 4, № 6. P. 926–935.
- D’hahan N. et al. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP // Proc. Natl. Acad. Sci. 1999. Vol. 96, № 21. P. 12162–12167.
- Saegusa N. et al. Kir6.2-deficient mice are susceptible to stimulated ANP secretion: KATP channel acts as a negative feedback mechanism? // Cardiovasc. Res. 2005. Vol. 67, № 1. P. 60–68.
- Flagg T.P. et al. Differential structure of atrial and ventricular KATP: Atrial KATP channels require SUR1 // Circ. Res. 2008. Vol. 103, № 12. P. 1458–1465.
- Fedorov V. V. et al. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts // J. Mol. Cell. Cardiol. 2011. Vol. 51, № 2. P. 215–225.
- Foster M.N., Coetzee W.A. KATP Channels in the Cardiovascular System // Physiol. Rev. 2016. Vol. 96, № 1. P. 177–252.
- Baron A. et al. A Novel KATP Current in Cultured Neonatal Rat Atrial Appendage Cardiomyocytes // Circ. Res. 1999. Vol. 85. P. 707–715.
- Van Wagoner D.R. Mechanosensitive gating of atrial ATP-sensitive potassium channels. // Circ. Res. 1993. Vol. 72, № 5. P. 973–983.
- Barajas-Martínez H. et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8 // Hear. Rhythm. 2012. Vol. 9, № 4. P. 548–555.
- Crotti L. et al. Spectrum and Prevalence of Mutations Involving BrS1- Through BrS12-Susceptibility Genes in a Cohort of Unrelated Patients Referred for Brugada Syndrome Genetic Testing // J. Am. Coll. Cardiol. 2012. Vol. 60, № 15. P. 1410–1418.
- Delaney J.T. et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation // Europace. 2012. Vol. 14, № 10. P. 1428–1432.
- Haïssaguerre M. et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel // J. Cardiovasc. Electrophysiol. 2009. Vol. 20, № 1. P. 93–98.
- Medeiros-Domingo A. et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac KATP channel Kir6.1 as a pathogenic substrate for J-wave syndromes // Hear.Rhythm. Elsevier Inc., 2010. Vol. 7, № 10. P. 1466–1471.
- Veeramah K.R. et al. The KCNJ8-S422L variant previously associated with J-wave syndromes is found at an increased frequency in Ashkenazi Jews // Eur. J. Hum. Genet. 2014. Vol. 22, № 1. P. 94–98.
- Miki T. et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1 // Nat. Med. 2002. Vol. 8, № 5. P. 466–472.
- Chutkow W.A. et al. Episodic coronary artery vasospasm and hypertension developed in the absence of Sur2 KATP channels // J. Clin. Invest. 2002. Vol. 110, № 2. P. 203–208.
- Daut J. et al. Hypoxic dilation of coronary arteries is mediated by ATP-sensitive potassium channels // Science (80-. ). 1990. Vol. 247, № 4948. P. 1341–1344.
- Duncker D.J. et al. Endogenous adenosine mediates coronary vasodilation during exercise after K+ATP channel blockade // J. Clin. Invest. 1995. Vol. 95, № 1. P. 285–295.
- Duncker D.J. et al. Role of K+ATP Channels in Coronary Vasodilation During Exercise // Circulation. 1993. Vol. 88, № 3. P. 1245–1253.
- Malester B. et al. Transgenic expression of a dominant negative KATP channel subunit in the mouse endothelium: effects on coronary flow and endothelin-1 secretion // FASEB J. 2007. Vol. 21, № 9. P. 2162–2172.
- Chutkow W.A. et al. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 KATP channels // J. Clin. Invest. 2002. Vol. 110, № 2. P. 203–208.
- Noma A. ATP-regulated K+ channels in cardiac muscle // Nature. 1983. Vol. 305. P. 147–148.
- Garlid K.D. Cation transport in mitochondria — The potassium cycle // Biochim. Biophys. Acta - Bioenerg. 1996. Vol. 1275, № 1–2. P. 123–126.
- Fryer R.M. et al. Ischemic preconditioning in rats: role of mitochondrial KATP channel in preservation of mitochondrial function // Am. J. Physiol. Circ. Physiol. 2000. Vol. 278, № 1. P. H305–H312.
- Oe K. et al. Modulation of norepinephrine release by ATP-dependent K+- channel activators and inhibitors in guinea-pig and human isolated right atrium // Cardiovasc. Res. 1999. Vol. 43, № 1. P. 125–134.
- Stanley C.A., Baker L. Hyperinsulinism in Infancy: Diagnosis by Demonstration of Abnormal Response to Fasting Hypoglycemia // Pediatrics. 1976. Vol. 57, № 5. P. 702–711.
- Thomas P.M. et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy // Science. 1995. Vol. 268, № 5209. P. 426–429.
- Nestorowicz A. et al. Mutations in the sulfonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews // Hum. Mol. Genet. 1996. Vol. 5, № 11. P. 1813–1822.
- Otonkoski T. et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland // Diabetes. 1999. Vol. 48, № 2. P. 408–415.
- Nestorowicz A. et al. Genetic Heterogeneity in Familial Hyperinsulinism // Hum. Mol. Genet. 1998. Vol. 7, № 7. P. 1119–1128.
- Thomas P., Ye Y., Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy // Hum. Mol. Genet. 1996. Vol. 5, № 11. P. 1809–1812.
- Nestorowicz A. et al. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism // Diabetes. 1997. Vol. 46, № 11. P. 1743–1748.
- Pinney S.E. et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations // J. Clin. Invest. 2008. Vol. 118, № 8. P. 2877–2886.
- Babenko A.P. et al. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus // N. Engl. J. Med. 2006. Vol. 355, № 3. P. 456–466.
- Yorifuji T. et al. The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitus // J. Clin. Endocrinol. Metab. 2005. Vol. 90, № 6. P. 3174–3178.
- Shimomura K. et al. A novel mutation causing DEND syndrome: A treatable channelopathy of pancreas and brain // Neurology. 2007. Vol. 69, № 13. P. 1342–1349.
- Shimomura K. et al. The first clinical case of a mutation at residue K185 of Kir6.2 (KCNJ11): A major ATP-binding residue // Diabet. Med. 2010. Vol. 27, № 2. P. 225–229.
- Edghill E.L. et al. Activating mutations in the KCNJ11 gene encoding the ATP-sensitive K+ channel subunit Kir6.2 are rare in clinically defined type 1 diabetes diagnosed before 2 years // Diabetes. 2004. Vol. 53, № 11. P. 2998–3001.
- Vedovato N. et al. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk // Diabetologia. Diabetologia, 2016. Vol. 59, № 7. P. 1430–1436.
- Shimomura K., Maejima Y. KATP Channel Mutations and Neonatal Diabetes // Intern. Med. 2017. Vol. 56, № 18. P. 2387–2393.
- Proks P. et al. Mechanism of action of a sulphonylurea receptor SUR1 mutation (F132L) that causes DEND syndrome // Hum. Mol. Genet. 2007. Vol. 16, № 16. P. 2011–2019.
- Proks P. et al. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes // Hum. Mol. Genet. 2006. Vol. 15, № 11. P. 1793–1800.
- Männikkö R. et al. A conserved tryptophan at the membrane-water interface acts as a gatekeeper for Kir6.2/SUR1 channels and causes neonatal diabetes when mutated // J. Physiol. 2011. Vol. 589, № 13. P. 3071–3083.
- Gloyn A.L. et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes // N. Engl. J. Med. 2004. Vol. 350, № 18. P. 1838–1849.
- Hani E.H. et al. Missense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): A meta-analysis suggests a role in the polygenic basis of Type II diabetes mellitus in Caucasians // Diabetologia. 1998. Vol. 41, № 12. P. 1511–1515.
- Souza S.W. et al. Polymorphism E23K (rs5219) in the KCNJ11 gene in Euro-Brazilian subjects with type 1 and 2 diabetes // Genet. Mol. Res. 2017. Vol. 16, № 2. P. 1–9.
- Sokolova E.A. et al. Replication of KCNJ11 (p.E23K) and ABCC8 (p.S1369A) association in Russian diabetes mellitus 2 type cohort and meta-analysis // PLoS One. 2015. Vol. 10, № 5. P. 1–21.
- Gloyn A.L. et al. Large-Scale association Studies of Variants in Genes Encoding the Pancreatic β-Cell KATP Channel Subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) Confirm That the KCNJ11 E23K Variant Is Associated with Type 2 Diabetes // Diabetes. 2003. Vol. 52, № February. P. 568–572.
- Schwanstecher C., Meyer U., Schwanstecher M. KIR6.2 Polymorphism Predisposes to T2D by Inducing Overactivity of β-Cell ATP-Sensitive K+ Channels // Diabetes. 2002. Vol. 51, № 3. P. 875–879.
- Carstensen B. et al. The E23K Variant of Kir6.2 Associates With Impaired Post-OGTT Serum Insulin Response and Increased Risk of Type 2 Diabetes // Diabetes. 2003. Vol. 52, № 2. P. 573–577.
- Florez J.C. et al. Haplotype structure and genotype phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. // Diabetes. 2004. Vol. 53, № May. P. 1360–1368.
- Hamming K.S.C. et al. Coexpression of the Type 2 Diabetes Susceptibility Gene ATP and Sulfonylurea Sensitivities of the ATP-Sensitive K+ Channel // Diabetes. 2009. Vol. 58, № 10. P. 2419–2424.
- Reyes S. et al. KATP channel Kir6.2 E23K variant overrepresented in human heart failure is associated with impaired exercise stress response // Hum. Genet. 2009. Vol. 126, № 6. P. 779–789.
- Reyes S. et al. KATP channel polymorphism is associated with left ventricular size in hypertensive individuals: A large-scale community-based study // Hum. Genet. 2008. Vol. 123, № 6. P. 665–667.
- Watanabe Y. et al. Electrophysiological analyses of transgenic mice overexpressing KCNJ8 with S422L mutation in cardiomyocytes // J. Pharmacol. Sci. 2017. Vol. 135, № 1. P. 37–43.
- Cooper P.E. et al. Cantú Syndrome Resulting from Activating Mutation in the KCNJ8 Gene // Hum. Mutat. 2014. Vol. 35, № 7. P. 809–813.
- Giudicessi J.R., Ackerman M.J. Potassium-channel mutations and cardiac arrhythmias - Diagnosis and therapy // Nat. Rev. Cardiol. 2012. Vol. 9, № 6. P. 319–332.
- Cantú J.M. et al. A distinct osteochondrodysplasia with hypertrichosis-Individualization of a probable autosomal recessive entity // Hum. Genet. 1982. Vol. 60, № 1. P. 36–41.
- Van Bon B.W.M. et al. Cantú syndrome is caused by mutations in ABCC9 // Am. J. Hum. Genet. 2012. Vol. 90, № 6. P. 1094–1101.
- Harakalova M. et al. Dominant missense mutations in ABCC9 cause Cantú syndrome // Nat. Genet. 2012. Vol. 44, № 7. P. 793–796.
- Brownstein C.A. et al. Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities – Support for the role of KATP channels in this condition // Eur. J. Med. Genet. Elsevier Masson SAS, 2013. Vol. 56, № 12. P. 678–682.
- McClenaghan C. et al. Glibenclamide reverses cardiovascular abnormalities of Cantu syndrome driven by KATP channel overactivity // J. Clin. Invest. 2020. Vol. 130, № 3. P. 1116–1121.
- Messenger A.G., Rundegren J. Minoxidil: mechanisms of action on hair growth // Br. J. Dermatol. 2004. Vol. 150, № 2. P. 186–194.
- Kaler S.G. et al. Hypertrichosis and congenital anomalies associated with maternal use of minoxidil // Pediatrics. 1987. Vol. 79, № 3. P. 434–436.
- Nguyen K.H., Marks J.G. Pseudoacromegaly induced by the long-term use of minoxidil // J. Am. Acad. Dermatol. Mosby, 2003. Vol. 48, № 6. P. 962–965.
- Bienengraeber M. et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating // Nat. Genet. 2004. Vol. 36, № 4. P. 382–387.
- Smeland M.F. et al. ABCC9-related Intellectual disability Myopathy Syndrome is a KATP channelopathy with loss-of-function mutations in ABCC9 // Nat. Commun. 2019. Vol. 10, № 1. P. 4457.
- Hagiwara S. et al. Blocking effects of barium and hydrogen ions on the potassium current during anomalous rectification in the starfish egg // J. Physiol. 1978. Vol. 279, № 1. P. 167–185.
- Oonuma H. et al. Inward Rectifier K+ Current in Human Bronchial Smooth Muscle Cells // Am. J. Respir. Cell Mol. Biol. 2012. Vol. 26, № 3. P. 371–379.
- Hagiwara S. Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish // J. Gen. Physiol. 1976. Vol. 67, № 6. P. 621–638.
- Jin W., Lu Z. A Novel High-Affinity Inhibitor for Inward-Rectifier K+ Channels // Biochemistry. 1998. Vol. 37, № 38. P. 13291–13299.
- Ramu Y., Xu Y., Lu Z. Engineered specific and high-affinity inhibitor for a subtype of inward-rectifier K+ channels // Proc. Natl. Acad. Sci. 2008. Vol. 105, № 31. P. 10774–10778.
- Takanari H. et al. Efficient and specific cardiac IK1 inhibition by a new pentamidine analogue // Cardiovasc. Res. 2013. Vol. 99, № 1. P. 203–214.
- Ji Y. et al. PA-6 inhibits inward rectifier currents carried by V93I and D172N gain-of-function KIR2.1 channels, but increases channel protein expression // J. Biomed. Sci. Journal of Biomedical Science, 2017. Vol. 24, № 1. P. 1–10.
- Swale D.R. et al. Article Computational and Functional Analyses of a Small-Molecule Binding Site in ROMK // Biophysj. Biophysical Society, 2015. Vol. 108, № 5. P. 1094–1103.
- Aréchiga-Figueroa I.A. et al. High-potency block of Kir4.1 channels by pentamidine: Molecular basis // Eur. J. Pharmacol. 2017. Vol. 815. P. 56–63.
- De Boer T. et al. The anti-protozoal drug pentamidine blocks KIR2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel // Br. J. Pharmacol. 2010. Vol. 159, № 7. P. 1532–1541.
- Imredy J.P., Chen C., MacKinnon R. A snake toxin inhibitor of inward rectifier potassium channel ROMK1 // Biochemistry. 1998. Vol. 37, № 42. P. 14867–14874.
- Wu M. et al. A potent and selective small molecule Kir2.1 inhibitor // Probe Reports from NIH Mol. Libr. Progr. 2010. P. 1–23.
- Kharade S. et al. Pore polarity and charge determine differential block of Kir1.1 and Kir7.1 potassium channels by the small-molecule inhibitor VU590 // Mol. Pharmacol. 2017. Vol. 37232. P. mol.117.108472.
- Lewis L.M. et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1 // Mol. Pharmacol. 2009. Vol. 76, № 5. P. 1094–1103.
- Bhave G. et al. Development of a Selective Small-Molecule Inhibitor of Kir1.1, the Renal Outer Medullary Potassium Channel // Mol. Pharmacol. 2011. Vol. 79, № 1. P. 42–50.
- Kharade S. V. et al. Discovery, Characterization, and Effects on Renal Fluid and Electrolyte Excretion of the Kir4.1 Potassium Channel Pore Blocker, VU0134992 // Mol. Pharmacol. 2018. Vol. 94, № 2. P. 926–937.
- Swale D.R. et al. ML418: The First Selective, Sub-Micromolar Pore Blocker of Kir7.1 Potassium Channels // ACS Chem. Neurosci. 2016. Vol. 7, № 7. P. 1013–1023.
- Scherer D. et al. Inhibition of inwardly rectifying Kir2.x channels by the novel anti-cancer agent gambogic acid depends on both pore block and PIP2 interference // Naunyn. Schmiedebergs. Arch. Pharmacol. Naunyn-Schmiedeberg’s Archives of Pharmacology, 2017. Vol. 390, № 7. P. 701–710.
- Zünkler B.J. et al. Cytosolic ADP enhances the sensitivity to tolbutamide of ATP-dependent K+ channels from pancreatic B-cells // FEBS Lett. 1988. Vol. 239, № 2. P. 241–244.
- Venkatesh N., Lamp S.T., Weiss J.N. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle // Circ. Res. 1991. Vol. 69, № 3. P. 623–637.
- Martin G.M. et al. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating // Elife. 2017. Vol. 6. P. 1–21.
- Martin G.M. et al. Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM // Elife. 2017. Vol. 6. P. 172908.
- Li N. et al. Structure of a Pancreatic ATP-Sensitive Potassium Channel // Cell. 2017. Vol. 168, № 1–2. P. 101-110.e10.
- M Ashcroft F. et al. New windows on the mechanism of action of KATP channel openers // Trends Pharmacol. Sci. 2000. Vol. 21, № November. P. 439–445.
- Yokoshiki H. et al. Levosimendan, a novel Ca2+ sensitizer, activates the glibenclamide-sensitive K+ channel in rat arterial myocytes // Eur. J. Pharmacol. 1997. Vol. 333, № 2–3. P. 249–259.
- Yokoshiki H. et al. The novel calcium sensitizer levosimendan activates the ATP-sensitive K+ channel in rat ventricular cells // J. Pharmacol. Exp. Ther. 1997. Vol. 283, № 1. P. 375–383.
- Kaheinen P. et al. Levosimendan increases diastolic coronary flow in isolated guinea-pig heart by opening ATP-sensitive potassium channels // J. Cardiovasc. Pharmacol. 2001. Vol. 37, № 4. P. 367–374.
- Wu S.N., Wu A.Z., Sung R.J. Identification of two types of ATP-sensitive K+ channels in rat ventricular myocytes // Life Sci. 2007. Vol. 80, № 4. P. 378–387.
- Paggio A. et al. Identification of an ATP-sensitive potassium channel in mitochondria // Nature. 2019. Vol. 572, № 7771. P. 609–613.
