
Движение против воли: гиперкинетический аспект
более аккуратная вертка статьи тут
это через время скроем.
Механизм формирования деятельности на уровне базальных ганглиев
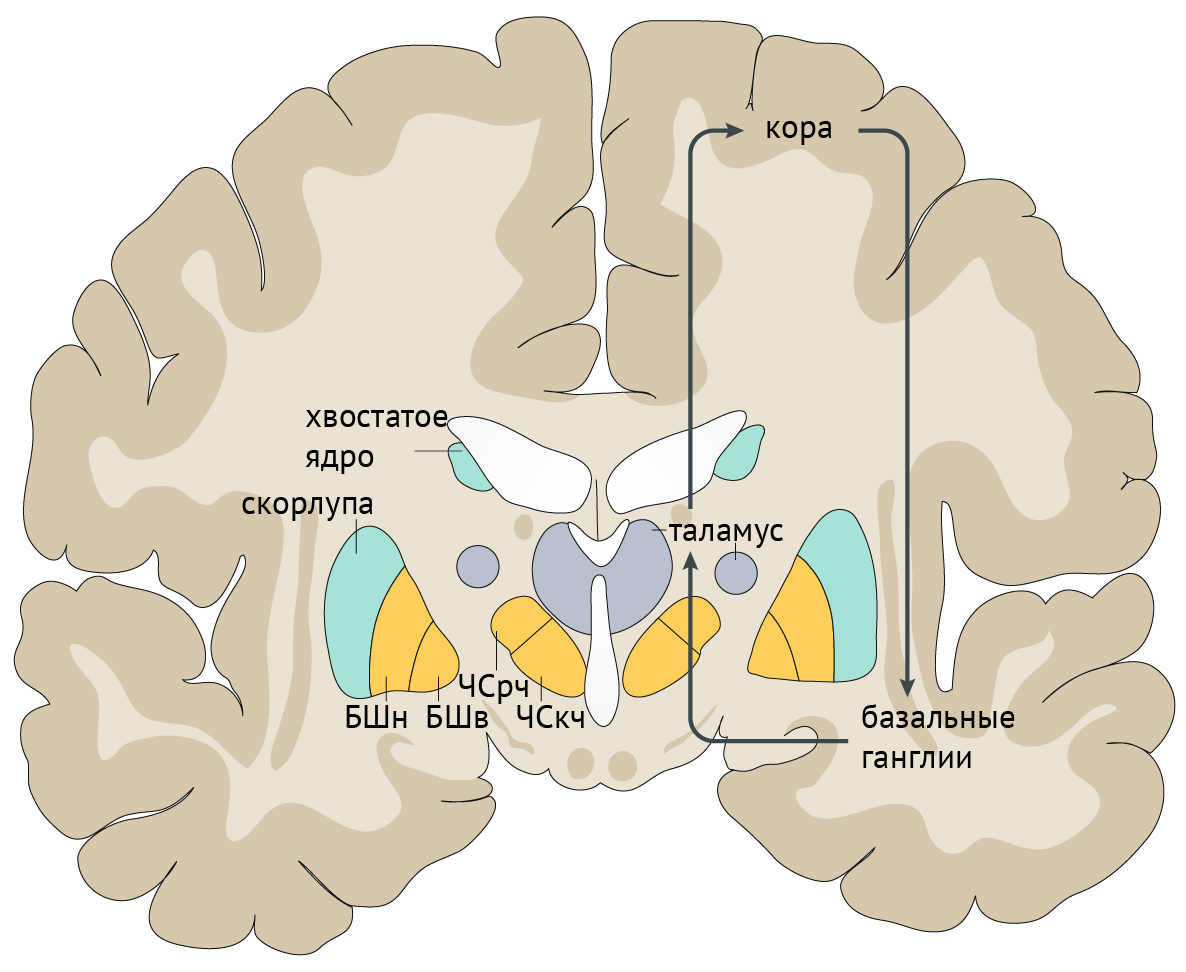
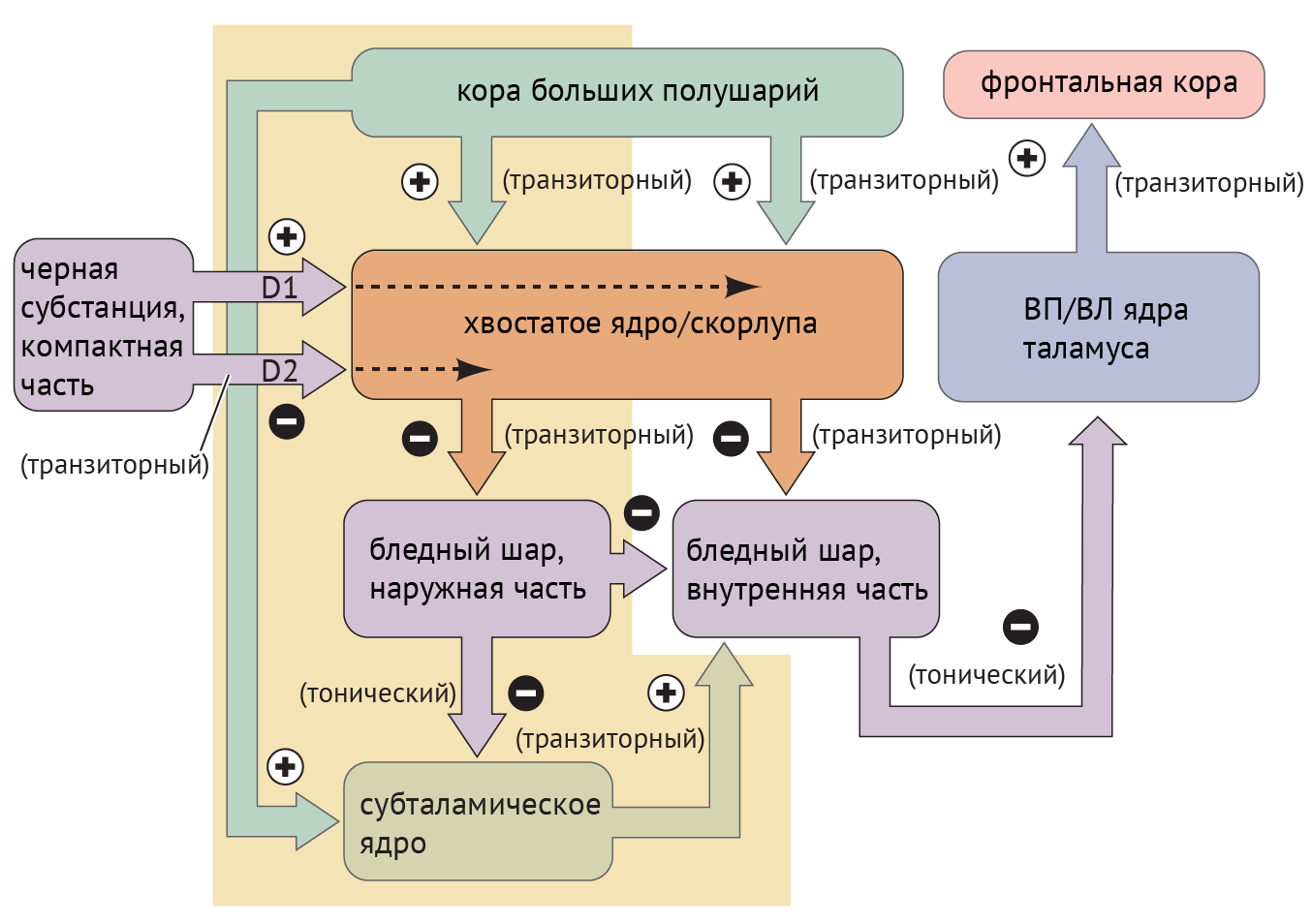
В рамках реализации программы экстрапирамидной системы наибольшее внимание отдается циклу «кора — базальные ганглии — таламус» (рис.1). Полосатое тело отправляет ГАМК-ергические сигналы к бледному шару (БШ), обусловливая растормаживание таламуса через прямой путь и снижая его активность непрямым путем — через внутренний сегмент бледного шара (БШв) и субталамическое ядро (СТЯ). Компактная часть черной субстанции (ЧСкч) обеспечивает стимуляцию дофаминовых рецепторов полосатого тела, активируя прямой путь через дофаминовые рецепторы D1 (D1R) и подавляя непрямой путь через дофаминовые рецепторы D2 (D2R) (схема 1).
Рассмотрим работу этих структур на примерах заболеваний, при которых симптомы экстрапирамидных нарушений выходят на первый план.
Болезнь Паркинсона
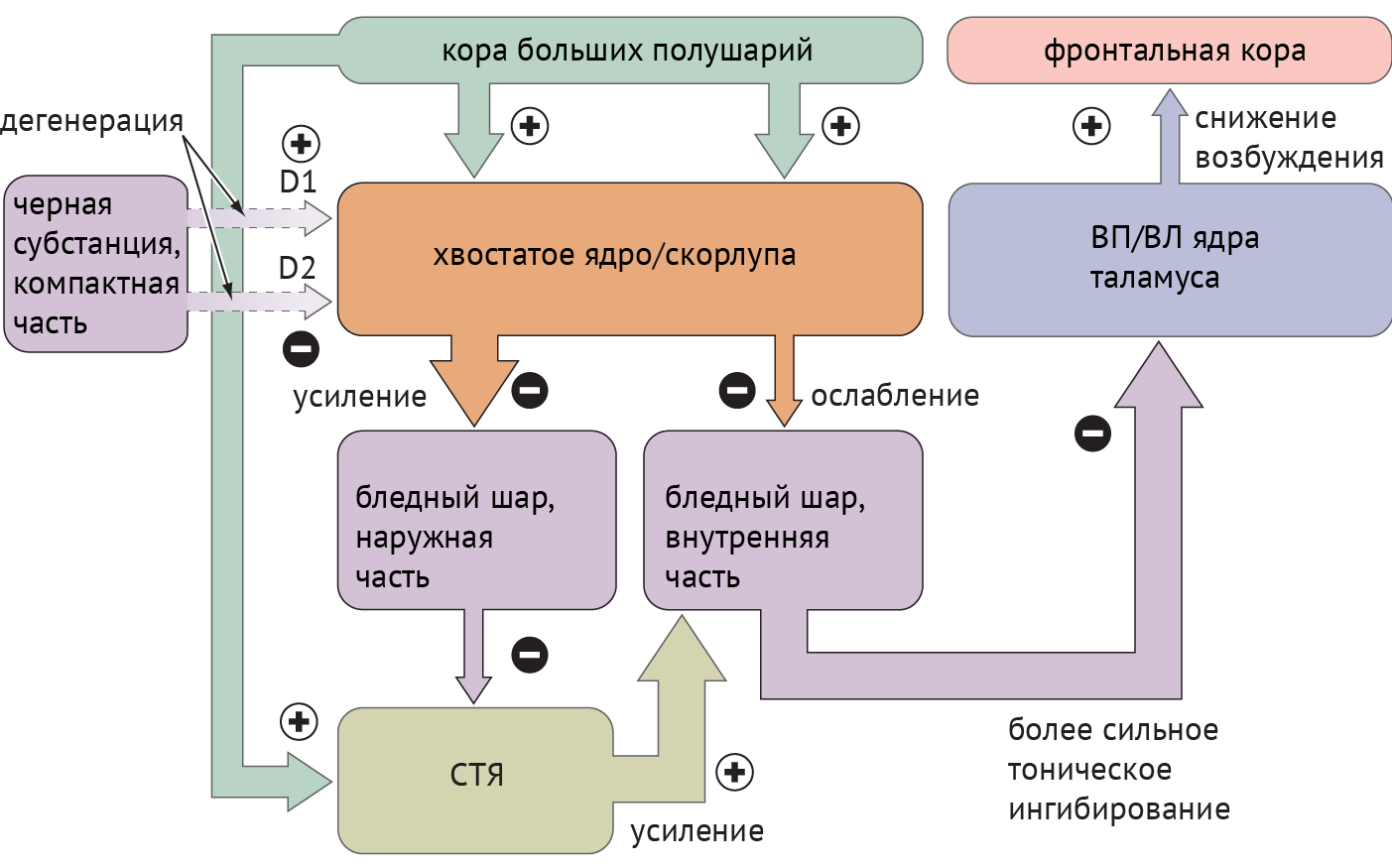
Схема 2. Схематическое отображение цикла «базальные ганглии — таламус — кора» при болезни Паркинсона [1]
Дегенерация ЧСкч приводит к недостаточной активации полосатого тела через прямой путь и, как следствие, дезингибированию БШв, который, в свою очередь, подает ингибирующие сигналы в таламус. Недостаточное ингибирование через непрямой путь ведет к подавлению БШн и дезингибированию СТЯ, которое активирует БШв (схема 2).
Болезнь Гентингтона
Следующий яркий представитель патологии базальных ганглиев — болезнь Гентингтона (Хантингтона).
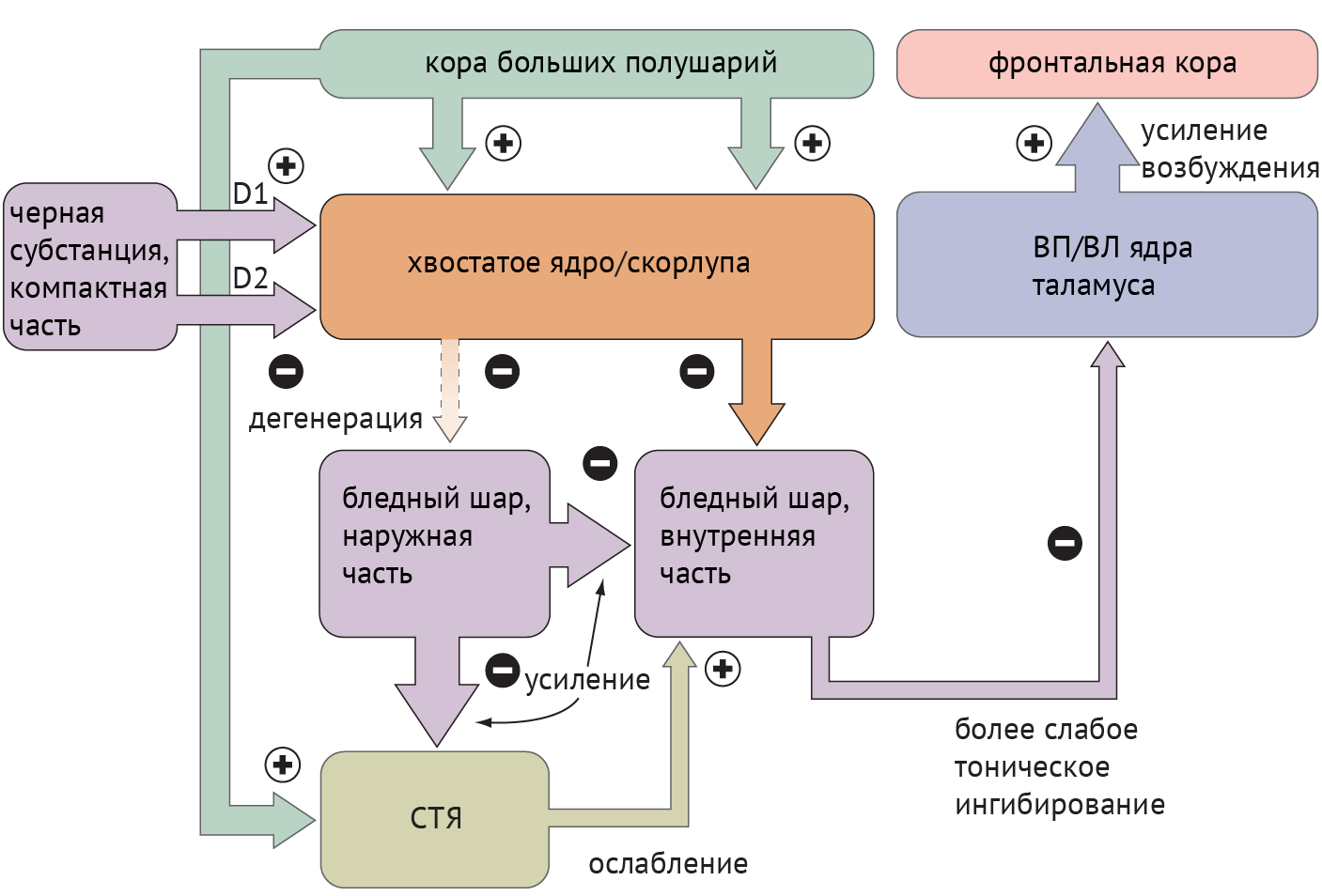
Схема 3. Схематическое изображение цикла «базальные ганглии — таламус — кора» при болезни Хантингтона [1].
Среди патологий, обсуждаемых в медицинских вузах, патогенез хореиформных гиперкинезов рассматривался следующим образом: локальная, но выраженная дегенерация шипиковых нейронов стриатума, проецирующихся на БШн, приводит к его дезингибированию, в результате чего угнетается активирующая деятельность СТЯ в отношении БШв и ингибиторная деятельность БШв в отношение таламуса (схема 3).
Если говорить о баллизме, то причиной развития этого синдрома будет являться поражение СТЯ. В таком случае, отсутствие стимуляции БШв со стороны СТЯ обусловливает недостаточную ингибирующую активность в отношении таламуса, что, в свою очередь, и реализуется в виде гиперкинеза.
Дистония — явление недостаточной ингибирующей активности, обусловленное поражением БШв и приводящее к дезингибированию таламуса и избыточной активации коры (схема 4).
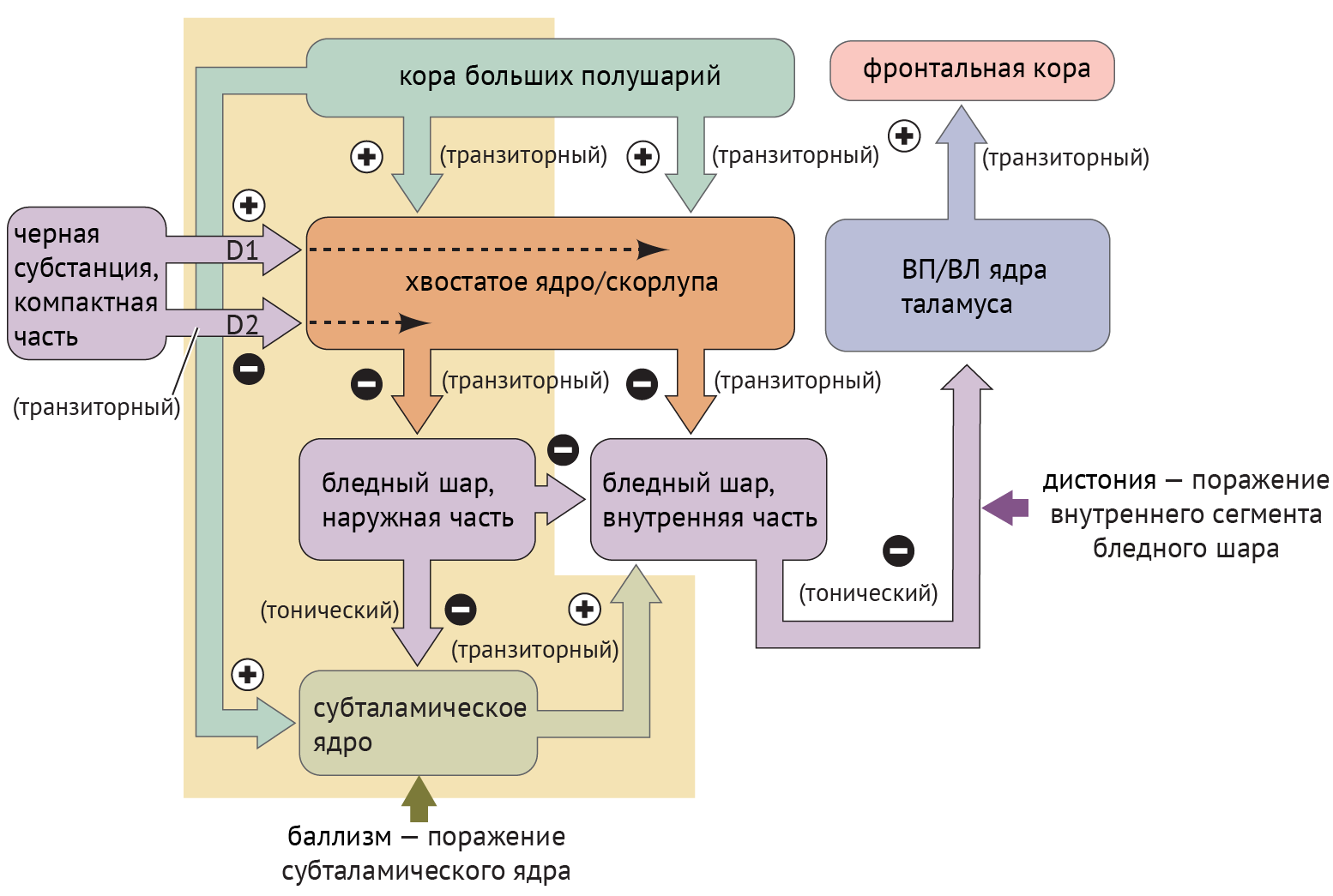
Схема 4. Схематическое изображение цикла «базальные ганглии — таламус — кора» при дистонии и баллизме [1].
Однако, недальновидным представлением о базальных ганглиях является убеждение, что гиперкинезы и гипокинезия обусловлены гипер- и гипофункцией этих структур. В таком случае можно поинтересоваться механизмом возникновения тремора при болезни Паркинсона (БП), а также почему стимуляция СТЯ снижает выраженность как дистонии, так и БП. Если так, то почему возможно развитие комбинированной дистонии с наличием симптомов паркинсонизма?
В самом деле, однозначно что-либо утверждать или поставить точку в исследованиях патогенеза экстрапирамидных расстройств на сегодняшний день еще невозможно. Однако имеющиеся в нашем распоряжении данные уже сейчас позволяют нам диагностировать эти патологии и по возможности вмешиваться в их течение.
Дистония
Дистония характеризуется аномальными непроизвольными движениями или позами, возникающими вследствие длительных или периодических сокращений мышц [2]. Наглядно это показано на иллюстрациях ниже (рис. 2).
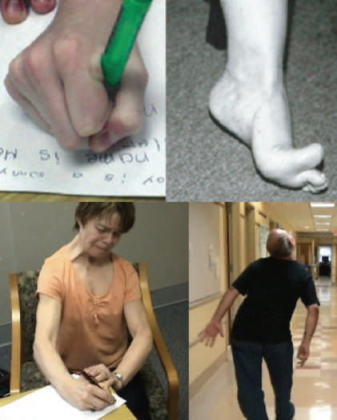
b) Стопа при дистонии.
c) Дистония рук, шеи и лицевых мышц, усиливающаяся во время письма. Заболевание манифестировало писчим спазмом, затем прогрессировало на мышцы шеи (тортиколлис) и мышцы лица.
d) Непроизвольное сгибание туловища и разгибание шеи (ретроколлис) во время походки.
В изучении этого феномена будет правильным, сперва, его классифицировать.
Классификация, определяющая, какую часть тела вовлекает дистония:
- Генерализованная дистония: дистония, вовлекающая торс и как минимум две другие части тела.
- Шейная дистония: дистония, вовлекающая шею и приводящая к формированию аномальных поз головы: тортиколлис (поворот головы в сторону), латероколлис (наклон головы в сторону), ретроколлис (разгибание шеи), антеколлис (сгибание шеи).
- Краниальная дистония: дистония, вовлекающая лицо или голосовые связки; может манифестировать как ларингеальная дистония (спастическая дисфония), затем блефароспазм, и позже как оромандибулярная дистония или комбинация последних двух (названная синдромом Мейжа).
- Ларингеальная дистония (названная спастической дисфонией): дистония, вовлекающая голосовые связки, приводя к удушью, к огрубению голоса с частыми изменениями высоты звука (аддукторный тип) или, что реже, к шепчещему, хриплому голосу (абдукторный тип).
- Блефароспазм: дистония, характеризующаяся спастическими закрываниями глаза.
- Оромандибулярная дистония: дистония, вовлекающая рот и/или челюсть, приводя к развитию самопроизвольных движений периоральной мускулатуры, открытию или закрытию рта.
- Гемидистония: дистония, вовлекающая только одну половину тела.
Дистонии, возникающие в результате определенных движений или действия триггеров:
- Писчий спазм: фокальная, специфическая дистония, вовлекающая руку и/или предплечье; проявляется во время попытки написать что-либо от руки, усиливаясь при продолжении письма.
- Спазм музыканта: специфическая дистония, вовлекающая ту часть тела, которая непосредственно задействована в игре на музыкальном инструменте (именно во время игры на нем).
- Пароксизмальная дистония: дистония, обусловленная определенными триггерами. Например:
• Дистония, индуцированная упражнениями: дистония, проявляющаяся после продолжительных тренировок, ограниченная задействованными в физических упражнениях конечностями. Наиболее типичной является дистония стопы, индуцированная длительной ходьбой.
• Пароксизмальная кинезиогенная дискинезия: дистония, ограниченная эпизодическим формированием дистонической позы в результате резких движений.
Этим классификация дистонии не ограничивается. Далее разберем такие формы дистонии, как изолированная и комбинированная.
Как понятно из названия, изолированная форма предполагает развитие у пациента симптомов одной только дистонии. Комбинированная — включает проявления других неврологических расстройств вдобавок к дистонии, при этом они вариабельны и зависят от этиологии основного заболевания.
Итак, непосредственно о причинах самого заболевания. Для этого взглянем на табл. 1.
Таблица. 1. Заболевания, одним из симптомов которых является дистония.
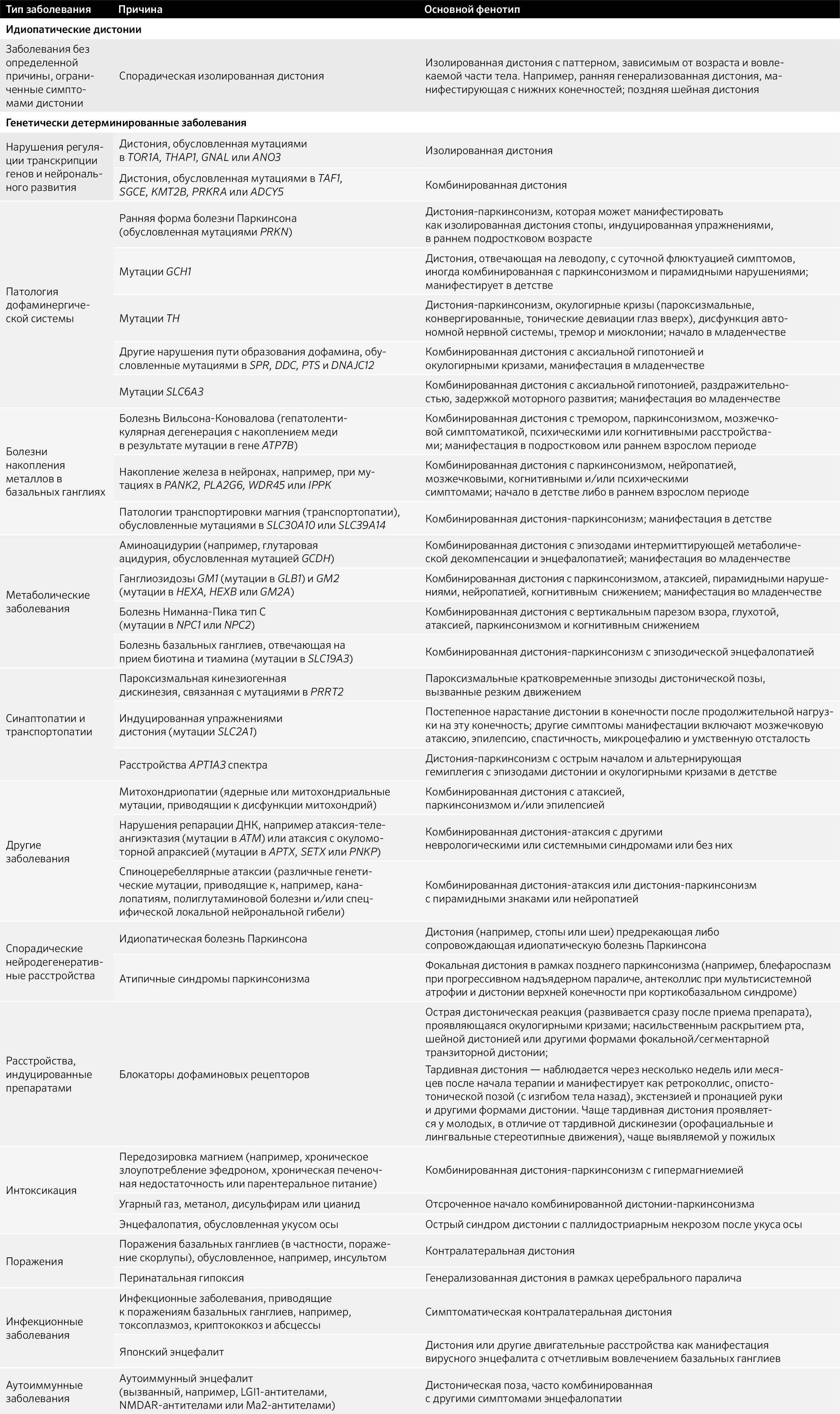
Среди дистоний описываются моногенные заболевания, проявляющиеся изолированной или комбинированной дистонией:
- Изолированные дистонии: аутосомно-доминантные — TOR1A (DYT1), THAP1 (DYT6) (редко аутосомно-рецессивное наследование), GNAL (DYT25) (редко аутосомно-рецессивное наследование), ANO3 (DYT24);
- Комбинированные дистонии:
а) аутосомно-доминантные — GCH1 (DYT5a) (редко аутосомно-рецессивное наследование), SGCE (DYT11), KMT2B (DYT28), ATP1A3 (DYT12), ADCY5 (редко аутосомно-рецессивное наследование);
б) аутосомно-рецессивные — TH (DYT5b), SPR, PRKRA (DYT16);
в) X-сцепленные — TAF1 (DYT3).
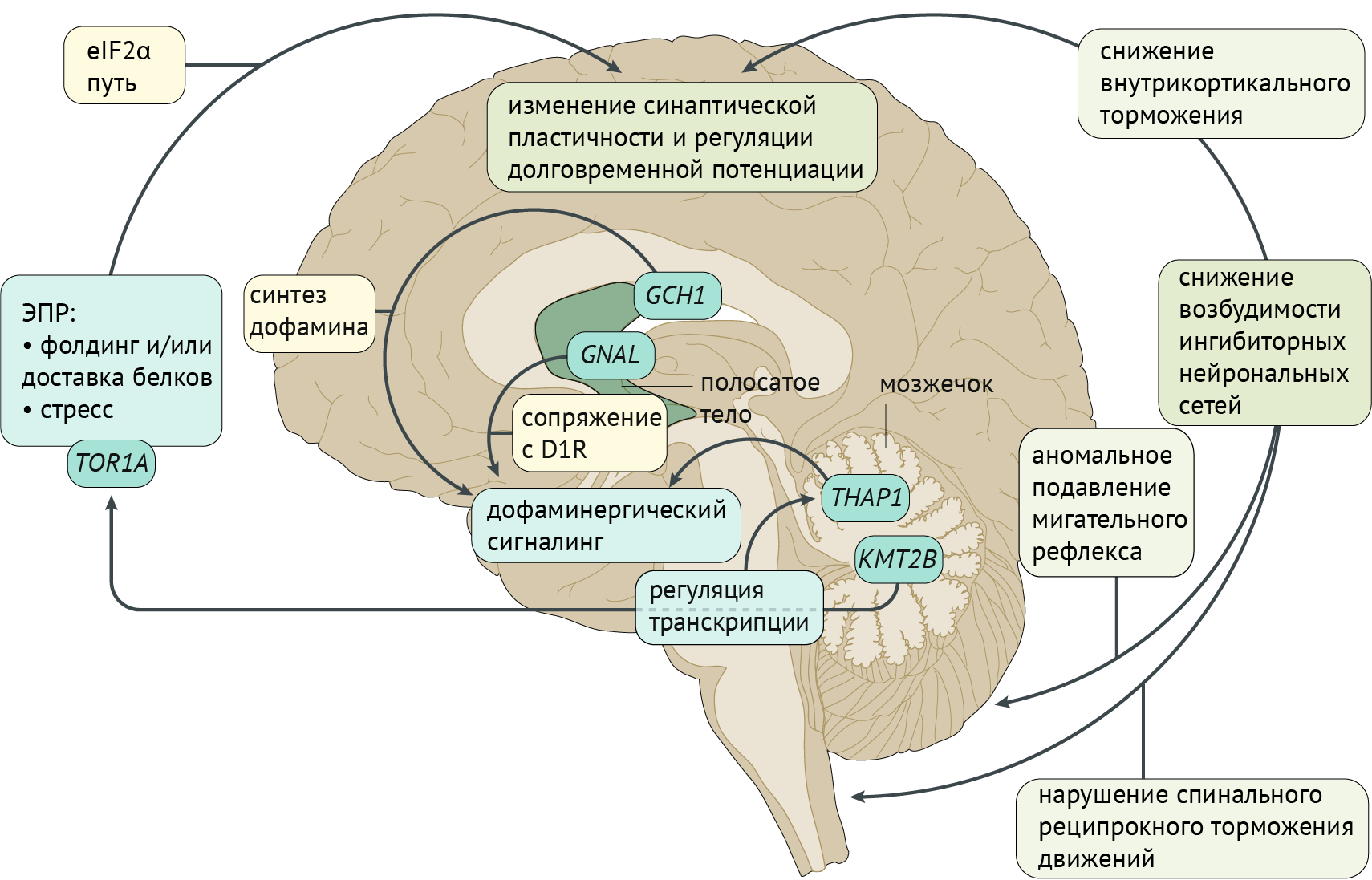
Рисунок 3. Патофизиология дистонии [2]
Патофизиология дистонии
Фундаментальная модель патофизиологии дистонии базируется на генетических и нейрофизиологических особенностях, изображенных на иллюстрации (рис. 3). Большинство генов, ассоциированных с дистонией, экспрессируются в стриатуме (GNAL, ANO3, ADCY5, HPCA и KCTD17), тогда как другие представлены в большей степени в мозжечке (THAP1 и KMT2B). Эти две структуры ЦНС играют значительную роль в патофизиологии дистонии. Молекулярная дисфункция, обусловленная мутациями этих генов, приводит к нарушениям в регуляции транскрипции генов, вовлеченных в регуляцию дофаминергической системы, а также в деятельности эндоплазматического ретикулума (ЭР). Фактор инициации трансляции эукариот 2α (eIF2α) связан с функционированием ЭР, модулируя синаптическую пластичность и долговременную потенциациию. В итоге наблюдается избыточная возбудимость коры, ствола и спинного мозга, что приводит к расстройствам движения (рис. 3).
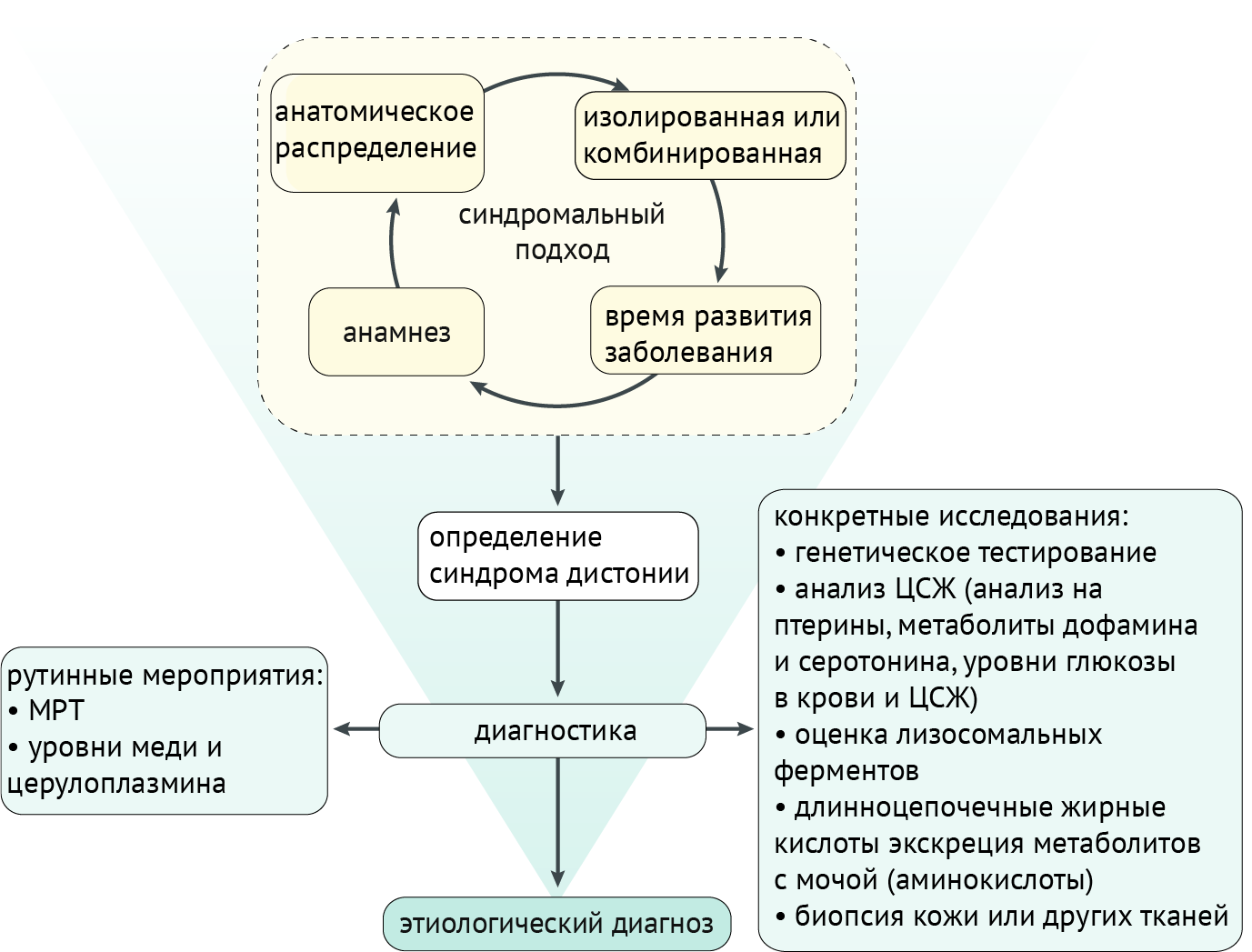
Диагностические мероприятия
Синдромальный подход, учитывающий историю и результаты клинического обследования (включая анатомическое распределение дистонии, возраст начала, развитие и наличие/отсутствие дополнительных неврологических или системных признаков), приводит к диагностике синдрома дистонии, что, в свою очередь, направляет дальнейшие поиски на выявление этиологии. Как правило, если клинические признаки соответствуют изолированной идиопатической дистонии, дальнейшие рутинные исследования направлены на исключение других патологий (которые редко могут проявляться изолированной дистонией): с использованием МРТ для исключения структурных нарушений или оценки уровней меди и церулоплазмина для скрининга болезни Вильсона-Коновалова. Если клинические особенности предполагают наличие другого заболевания, диагностическое обследование будет адаптировано к определенному пациенту, и потребуются более конкретные исследования, включая генетическое тестирование, анализ спинномозговой жидкости (ЦСЖ), биопсию тканей и т. д. (схема 5).
Красные флажки в диагностике идиопатических дистоний:
- Необычный паттерн клинической манифестации с учетом возраста, начала заболевания и вовлечения частей тела;
- Внезапное начало и быстрое прогрессирование;
- Перинатальная травма, нарушение развития;
- Фармакологический анамнез (например, прием блокаторов дофаминовых рецепторов);
- Наличие других неврологических или системных расстройств (которые бы указывали на комбинированную дистонию);
- Бульбарные расстройства, в т. ч. протрузия языка и дисфагия;
- Гемидистония (можно предположить структурное повреждение ЦНС, вызвавшее дистонию);
- Фиксированная дистония (почти всегда имеет психогенную этиологию, реже встречается при нейродегенеративных заболеваниях; вовлеченная в дистонию часть тела не возвращается в первоначальное состояние в покое, отсутствуют типичные для дистонии сенсорные трюки, нет вовлечения соседних мышц).
Сенсорные трюки
Пациенты с различными видами фокальной дистонии могут испытывать временное облегчение симптомов при выполнении определенных маневров, таких как касание подбородка, щеки, затылка при шейной дистонии; глазных яблок при блефароспазме; пишущей руки при писчем спазме. Подобные «сенсорные трюки» являются специфичными для фокальной дистонии симптомами. Трюки могут быть как тактильными, так и проприоцептивными. Даже мысленно представляя проделывание сенсорного трюка, пациент может облегчить выраженность мышечного спазма (рис. 4).
Также характерным феноменом, но не специфичным, может считаться наличие зеркальных движений. Например, при писчем спазме — письмо недоминантной рукой, не подверженной фокальной дистонии, провоцирует дистонию в другой руке [5]. Вгляните.
«Нулевая позиция» — характерное для дистонического тремора поза, в которой происходит регресс тремора. Используется для дифференциальной диагностики с эссенциальным тремором, при котором такой позиции не наблюдается. Эта диагностика особенно важна, когда в начале заболевания дистония не проявляет себя никак, кроме как тремором [5].
Терапия
Чаще всего терапия заключается в купировании выраженности дистонии. Конечно, если есть возможность вылечить заболевание, в первую очередь, терапия должна быть направлена на устранение его причины. Однако здесь мы поговорим именно о симптоматической и патогенетической терапии (схема 6).
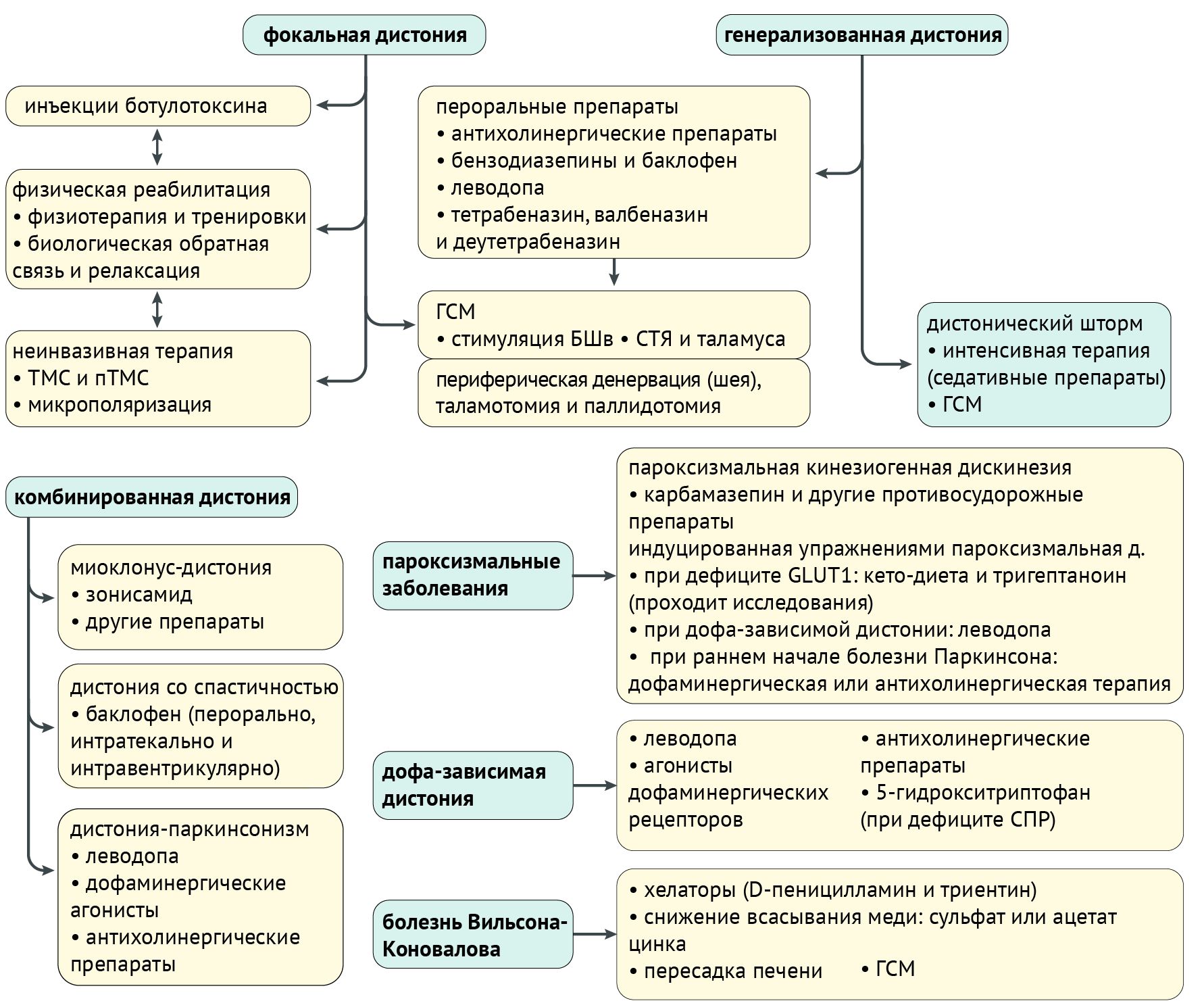
(ГЛЮТ1), глюкозный транспортер 1 типа эритроцитарный/мозговой; (БШв), бледный шар, внутренний сегмент; (пТСМ), повторная транскраниальная магнитная стимуляция; (СПР), сепиаптерин редуктаза; (СТЯ), субталамическое ядро; (тСПТ), транскраниальная стимуляция постоянным током; (ТМС), транскраниальная магнитная стимуляция.
С целью оценки эффективности терапии можно пользоваться следующими шкалами: Шкалой Бурке-Фана-Марсдена, Унифицированной рейтинговой шкалой дистонии (UDRS), Рейтинговой шкалой спастической кривошеи западного Торонто (TWSTRS), Шкалой влияния шейной дистонии на качество жизни (CDIP-58), Опросником по краниоцервикальной дистонии (CDQ-24) и множество других шкал, используемых отдельно для писчего спазма, оромандибулярной, торсионной дистонии и т. д.
Ботулинотерапия
Механизм действия следующий: эндоцитоз ботулотоксина периферическими холинергическими терминалями приводит к нарушению функции синаптической трансмиссии через разрушение растворимого N-этилмалеимид-чувствительного фактора активации белкового рецептора (SNARE), необходимого для фузии везикул, содержащих нейромедиатор, через пресинаптическую мембрану. Ботулинотерапию используют в лечении почти всех форм фокальной и сегментарной дистонии и многих других заболеваний. Токсин вводят непосредственно в мышцу, обусловливающую дистоническое сокращение или движение. Улучшение обычно наступает в течение нескольких дней и длится до 3–4 месяцев, после чего инъекции повторяются. Большинство побочных эффектов носят транзиторный характер и ассоциированы с фокальной мышечной слабостью, включая птоз у пациентов с блефароспазмом или слабость мышц шеи и дисфагию у пациентов с цервикальной дистонией.
Физиотерапия и вспомогательные методы
Физиотерапия в первую очередь направлена на улучшение осанки, двигательной функции и профилактики контрактур. Такая терапия наиболее эффективна при наличии синергических дистонических паттернов, вовлекающих запястье и предплечье (комбинирование пронации или супинации, сгибания или разгибания) нежели пальцы кисти (сгибание пальцев). К физиотерапии также относятся и вышеописанные сенсорные трюки. Облегчить дистонию, кроме того, позволяют различные девайсы и приспособления.
Вспомогательные методы включают в себя неинвазивные стимуляционные техники, такие как повторная транскраниальная магнитная стимуляция (пТМС) и транскраниальная стимуляция постоянным током (тСПТ). Несмотря на воодушевляющие результаты, рутинное использование данных методов пока еще не представляется возможным.
Фармакотерапия
1. Антихолинергические препараты. Наибольшей эффективностью обладают при терапии генерализованных и сегментарных форм, нежели при фокальной дистонии. Тригексифенидил и другие антихолинергические средства наиболее действенны среди пациентов с изолированной генерализованной дистонией в сравнении с фокальной дистонией взрослых или комбинированной дистонией. Эти препараты также могут быть полезны при острой дистонической реакции или тардивной дистонии.
Побочные явления включают затуманенное зрение, сухость во рту, нарушение мочеиспускания, констипацию и когнитивное снижение. Эти явления наиболее характерны для возрастных пациентов, хотя среди больных с генерализованной дистонией чаще встречаются молодые пациенты. Так или иначе, эта группа препаратов лучше переносится детьми, среди которых побочные явления встречаются куда реже. Начало лечения с минимальной терапевтической дозы и постепенное титрование в течение нескольких недель позволит минимизировать риск развития побочных эффектов.
2. Леводопа. Препарат выбора в лечении дофамин-чувствительной дистонии. Хотя некоторые пациенты и могут испытывать тошноту, сонливость, головокружение и другие острые побочные эффекты, связанные с леводопой, в большинстве случаев не проявляется таких негативных явлений как двигательные флюктуации или дискинезии, типичные в случае приема леводопы пациентами с БП. Более того, пациенты с дофамин-чувствительной дистонией, как бы это ни звучало, более чувствительны к леводопе, нежели пациенты с БП — первым требуются меньшие дозы для достижения клинического улучшения. Использование леводопы при других формах дистонии практически бесполезно.
3. Антидофаминергические препараты: действуют в роли ингибиторов везикулярного транспортера моноаминов 2 (VMAT2). Тетрабеназин, дететрабеназин и валбеназин используются в терапии хореи, тиков и тардивной дискинезии, а также демонстрируют клиническую эффективность среди пациентов с дистонией, в частности, тардивной дистонии. Такие побочные явления, как сонливость, паркинсонизм, депрессия и акатизия, могут ограничить возможность использования этих лекарств, в связи с чем необходим тщательный мониторинг. Блокаторы дофаминовых рецепторов (например, галоперидол и пимозид), наоборот, могут привести к тардивной дискинезии, из-за чего не рекомендуются к использованию в терапии дистонии.
4. Баклофен: агонист ГАМКB-рецептора, снижает выраженность и частоту дистонических движений у некоторых пациентов с дистонией (в частности при сочетании оромандибулярной дистонии с церебральным параличом, дистонии и хореоатетоза с гипертонусом мышц). Интратекальное и интравентрикулярное введение демонстрирует лучший результат, нежели пероральный прием баклофена. Использование баклофена является предпочтительном выбором для пациентов с церебральным параличом, а интравентрикулярная баклофеновая помпа при рефрактерной генерализованной дистонии у детей рассматривается как один из вариантов основной терапии. Однако данный аспект осложняется возможным развитием нейроинфекции, связанной с миграцией или смещением катетера.
5. Другие препараты. Бензодиазепины (такие как диазепам, клоназепам, лоразепам и мидазолам) могут использоваться в качестве офф-лейбл терапии мышечной дистонии, однако их применение ограничено побочными эффектами, основные из которых сонливость и аддикция.
- Другие мышечные релаксанты также продемонстрировали эффективность у пациентов с сегментарной или генерализованной дистонией. Среди таковых циклобензаприн, орфенадрин и хлорзоксазон.
- Зонизамид — антиконвульсант, блокатор натрий- и кальций-зависимых рецепторов и моноаминоксидазы, который проявляет эффективность в отношении миоклонус-дистонии.
- Золпидем связывается с ГАМКА-рецептор и модулирует его, удлиняя время открытия. Препарат эффективен при различных типах генерализованной дистонии.
- Габапентин, аналог ГАМК, действует как α2δ лиганд кальциевого канала, и также может быть полезен в терапии.
- Топическое использование клонидина (агониста α2-адренорецепторов) в виде глазных капель находит свое применение у пациентов с блефароспазмом.
Хирургическое лечение
1. Глубокая стимуляция мозга. ГСМ признается наиболее эффективным методом терапии при рефрактерной, инвалидизирующей дистонии (как при генерализованной, так и при тяжелой сегментарной и фокальной формах). Мишенью стимуляции являются БШв и СТЯ.
К побочным явлениям при стимуляции БШ относятся: дизартрия, паркинсонизм (включая нарушение походки, микрографию и брадикинезию). При стимуляции СТЯ: хорея, нарушение чувствительности, письма, снижение массы тела, дизартрия, боль в плече и депрессия. Общие побочные эффекты включают дискоординацию, парестезии и ощущение покалывания во рту, а также эффекты связанные с хирургической имплантацией и смещением датчика, инфицирование и образование гематомы.
Что интересно, ГСМ БШ демонстрирует наилучший эффект при дистониях, ассоциированных с мутациями TOR1A, KMT2B, SGCE, THAP1 и GNAL, хотя в случае последних двух эффективность все же вариабельна.
2. Абляция. Паллидотомия и таламотомия ушли в прошлое из хирургической практики, уступив место ГСМ.. Хотя, стоит отдать этому методу должное. К примеру, при длительном наблюдении за 15 пациентами с дистонией музыканта, среди 93 % пациентов, перенесших вентрооральную таламотомию, наблюдалось выраженное улучшение сразу после процедуры, которое поддерживалось в среднем около 31 месяца [7].
Дистонический шторм
Дистонический шторм, или дистонический статус — жизнеугрожающее, редкое состояние, являющееся осложнением генерализованной дистонии и обусловленное инфекцией, лихорадкой, прерыванием терапии, хирургическим вмешательством или травмой. Чаще встречается среди детей. Пациенты испытывают учащение и утяжеление эпизодов формирования болезненной вынужденной позы или иррегулярных мышечных дистонических подергиваний, которые могут быть ассоциированы с лихорадкой, респираторным дистрессом, дизавтономией (включая тахикардию, диафорез), рабдомиолизом и почечной недостаточностью. Дистонический статус требует экстренного медицинского вмешательства. Необходимо исключить триггеры (если причиной развития стала инфекция, назначить соответствующую терапию), также назначить моно- или политерапию с целью смягчения выраженности дистонии. Антихолинергические препараты, тетрабеназин, клонидин и баклофен должны быть использованы; внутривенное введение мидазолама, пропофола и недеполяризующих миорелаксантов можно рассматривать как дополнительные инструменты купирования состояния. Глубокая стимуляция мозга и интратекальная баклофеновая помпа также могут рассматриваться как эффективный метод купирования дистонического шторма. Отсутствие или несвоевременное начало медицинской помощи может привести к летальным последствиям.
Хорея
Пойдем по тому же пути, что и ранее. Что такое и как выглядит хорея?
«Хорея» в переводе с греческого означает танец. Хореиформные гиперкинезы представляют из себя насильственные, беспорядочные, быстрые движения конечностей, лица и головы. Пациенты с хореей пытаются замаскировать эти движения под свои собственные — этот феномен зовется «паракинезией». Другой характеристикой хореиформных гиперкинезов является неспособность формирования нормальных движений. Например, пациент с хореей Сиденгама, при попытке пожать человеку руку, вместо нормального рукопожатия будет совершать постепенно сжатие пальцев сверху вниз, подобное тому, как доят вымя («рука доярки»). При наличии хореи в оробукколингвальной области пациент не может держать высунутым язык более нескольких секунд.
Итак, основные характеристики хореи — непредсказуемость, неспособность производить нормальные движения, паракинезии [8].
Некоторые авторы считают баллизм частью хореиформных движений, отмечая схожесть и частое сочетание этих состояний и выделяя баллизм как тяжелую форму проксимальной хореи. Однако в отличие от хореи, которая чаще локализуется в средних и дистальных отделах конечностей, баллизм проявляется проксимально и характеризуется размашистыми, высокоамплитудными движениями, что, по мнению некоторых авторов, отделяет его от хореи [9].
В определенных нейрофизиологических аспектах эти явления схожи: при хорее происходит дезингибирование БШн и подавления активности СТЯ и БШв. Баллизм возникает при непосредственном повреждении СТЯ.
Атетоз — еще одно состояние, которое совместно с хорей и баллизмом относят к хореоформному спектру. Атетоз характеризуется медленными, извивающимися движениями в дистальных отделах конечностей, хотя может вовлекать и язык.
Но и на этот счет имеются противоречия: некоторые авторыпридерживаются позиции о принадлежности атетоза к дистониям. Трудно отличить атетоз от дистонической позы руки, при которой формируются плавные, медленные движения пальцев, в результате чего такие движение называют дистоническими хореоатетоидными. Такие движения встречаются при церебральном параличе [10].
Скорость двигательного компонента при хорее выше, чем при дистонии, но ниже, чем подергивания при миоклонии (0,2 сек). Кроме того, миоклонические подергивания можно спровоцировать обыкновенным постукиванием неврологическим молоточком по подверженным мышечным волокнам.
От тиков хорею отличают, как уже было сказано, отсутствие каких-либо строгих паттернов движений. Для тиков характерны определенные топографические локализации, частичная волевая компенсация, наличие триггеров и возможности предугадывания начала.
Стереотипии, наблюдаемые при анти-NMDA-энцефалите или тардивной дискинезии с вовлечением оробукколингвальной области, могут обладать сходной с хореей симптоматикой. Однако наличие стереотипного паттерна, повторяющегося вновь и вновь, как раз и отличает стереотипии от непредсказуемой хореи.
Крупноразмашистый, высокоамплитудный тремор при мозжечковой патологии также может вводить в заблуждение при диагностике хореиформных гиперкинезов. Однако тремор с элементом вращения вокруг оси, а также его активизация в движении (наличие постурального/кинетического, интенционного компонента) делают дифференциальную диагностику проще.
Наследственные хореи
Аутосомно-доминантные
Болезнь Хантингтона
Причина: повторы CAG в гене HTT, обуславливающие появление аномального белка HTT (хантингтина).
36–39 повторов — сниженная пенетрантность; > 40 — полная пенетрантность; > 60 — ювенильная форма (синдром Вестфаля)
Проявления: Хореиформные гиперкинезы с вовлечением верхней краниальной области, психиатрическая симптоматика (тревожность, депрессия, обсессивно-компульсивное расстройство), когнитивные нарушения.
Увеличение латентного периода саккад, аномалии в проведение теста антисаккад (исследователь просит уводить взгляд в противоположную сторону от движущегося объекта, например, пальца). Неспособность производить нормальные движения (протрузия языка и «рука доярки»). Коленный рефлекс по типу маятника (удар, или его имитация, молоточком вызывает маятникообразное покачивание ноги в коленном суставе).
При длительном течении заболевания паркинсонизм выходит на первый план.
Характерная походка по типу пританцовывания.
Дети: паркинсонизм, дистония и эпилептические припадки (синдром Вестфаля); хорея не характерна.
C9orf72-ассоциированные заболевания
Причина: GGGGCC повторы в гене C9orf72
Средний возраст начала — 43 года.
Проявления: Фенотипические проявления вариабельны: фронто-темпоральная деменция и боковой амиотрофический склероз, хорея, паркинсонизм, дистония, атаксия и миоклонус. Также может встречаться слабовыраженное когнитивное снижение. Наиболее встречаемое схожее с БХ заболевание.
Спиноцеребеллярная атаксия 17 типа (SCA-17)
Причина: CAG/CAA повторы в гене TBP (TATA-box связывающие белки)
Второе название — Хантингтон-подобное заболевание 4 типа.
Проявления: Атаксия, дистония, хорея, когнитивное снижение, психиатрические расстройства.
Второе по встречаемости фенотипически имитирующее БХ заболевание после C9orf72-связанных заболеваний.
Спиноцеребеллярная атаксия 1 типа (SCA-1)
Причина: Аномальная экспансия CAG в гене ATXN1. Симптоматика обычно проявляется при > 39 повторов
Проявления: Прогрессирующая мозжечковая атаксия, дизартрия, иногда вовлечение бульбарных функций. В начале заболевания — нарушение походки, невнятная речь, проблемы с равновесием, оживленные глубокие сухожильные рефлексы, гиперметрические саккады, нистагм и легкая дисфагия. Более поздние признаки включают снижение скорости саккад, вертикальный паралич взора вверх, дисметрию, дисдиадохокинез и мышечную гипотонию. На поздних стадиях наблюдаются мышечная атрофия, снижение глубоких сухожильных рефлексов, потеря проприоцепции, когнитивные нарушения (по типу лобной деменции), хорея, дистония и бульбарные расстройства.
Начало обычно в 30–40 лет, хотя сообщается о случаях заболевания среди детей и лиц пожилого возраста. Манифестация заболевания после 60 лет может проявляться только мозжечковой симптоматикой. Летальный исход наступает через 10–30 лет от начала заболевания; у лиц с ювенильным дебютом наблюдается быстрое прогрессирование и более тяжелое течение.
Сенсорная аксональная нейропатия, обнаруженная с помощью электрофизиологического тестирования, является распространенным явлением; нейровизуализация обычно демонстрирует атрофию мозжечка и ствола мозга [11].
Спиноцеребеллярная атаксия 2 типа (SCA-2)
Причина: Аномальная экспансия CAG в гене ATXN2. Фенотипически проявляется при количестве повторов > 33
Проявления: Характеризуется прогрессирующей мозжечковой атаксией, включая нистагм, медленные саккады, а у некоторых людей выявляется офтальмоплегия или паркинсонизм, реже — хорея. Присутствует пирамидная симптоматика. Глубокие сухожильные рефлексы оживлены на ранней и отсутствуют на поздней стадии заболевания.
Манифестирует обычно в 40 лет, продолжительность заболевания 10–15 лет с последующим летальным исходом [12].
Спиноцеребеллярная атаксия 3 типа (SCA-3)
Причина: Аномальная экспансия CAG в гене ATXN3.
Норма: 12–44 повторов;
промежуточное состояние: 45–59 повторов;
полная пенетрантность: 60–87 повторов
Наиболее встречаемое заболевание среди всей группы спиноцеребеллярных атаксий.
Проявления: прогрессирующая мозжечковая атаксия; пирамидная симптоматика; экстрапирамидная симптоматика, в основном проявляющаяся дистонией и ригидностью, реже — хорея; выраженная периферическая амиотрофия и генерализованная арефлексия; прогрессирующая наружная офтальмоплегия; фасцикуляции лицевых мышц и языка; экзофтальм; когнитивные, психиатрические нарушения и дизавтономия.
Начало заболевания приходится на возраст 20–50 лет и зависит от частоты тринуклеотидных повторов. Продолжительность жизни после манифестации заболевания от 6 до 29 лет [13].
Хантингтон-подобное заболевание 1 типа
Причина: Мутации гена кодирующего прионный белок на 20 хромосоме
Проявления: Хорея, атетоз, атаксия, когнитивные и психиатрические нарушения, эпилепсия. Манифестация в молодом и среднем возрасте. Заболевание фатально.
Хантингтон-подобное заболевание 2 типа
Причина: CTG-CAG тринуклеотидный повтор в гене JPH3 (junctophilin 3) на 16 хромосоме
Проявления: Хорея, паркинсонизм, снижение массы тела, деменция, психиатрические расстройства.
Дебют заболевание в среднем возрасте. Обычно через 20 лет после манифестации больные погибают.
Встречается практически только в африканской группе.
В 10 % случаев наблюдается акантоцитоз, но наследственный паттерн и нормальные уровни креатинфосфокиназы отличают заболевание от нейроакантоцитоза.
Дентато-рубро-паллидо-льюисова атрофия
Причина: Аномальная экспансия CAG в гене ATN1 на 12 хромосоме
Проявления: Для ювенильной формы (младше 20 лет) характерны мозжечковая атаксия, миоклонус, эпилепсия, прогрессирующие когнитивные расстройства. Для старшего возраста — атаксия, хореоатетоз, деменция и психиатрическая симптоматика.
Средний возраст начала 31 год.
Заболевание встречается среди дальневосточной азиатской (Япония, Корея, Сингапур) и португальской (Португалия, Латинская Америка) групп [14].
Нейроферритинопатия
Причина: Мутация FTL (легких цепей ферритина)
Проявления: Дистония с преимущественным поражением нижней краниальной области.
Низкий уровень сывороточного ферритина (не всегда)
МРТ в режимах GRE или SWI демонстрирует кистозную дегенерацию хвостатого ядра и скорлупы.
«Выделение карандашом» — корковые борозды будто выделены карандашом; связано с кумуляцией железа [15].
Доброкачественная врожденная хорея
Причина: Мутации в NKX2-1 (TITF-1)
Проявления: Манифестация заболевания приходится на младенческий возраст или раннее детство. Обычно прогрессирует минимально либо остается на плато с начала манифестации.
Атаксия, дистония, гипотония, хорея. Когнитивные функции обычно сохранны, но могут проявляться невыраженные нарушения. Кроме того, NKX2-1 задействован в функционировании легких и щитовидной железы. В 30–40 % случаев развивается синдром «мозг — легкие — щитовидная железа». В этом случае могут выявляться неонатальный респираторный дистресс-синдром, интерстициальная болезнь легких, врожденный гипотиреоидизм или гипоплазия щитовидной железы.
В целом, «доброкачественность» зачастую относится только к патологии ЦНС, хотя встречающиеся случаи когнитивных расстройств наталкивают на переосмысление «доброкачественности» даже в данном контексте. А учитывая, что некоторые пациенты страдают от частых легочных инфекций, легочного фиброза, злокачественных новообразований легких, мочевого пузыря и лейкемии, понятие «доброкачественности» в данном случае не применимо.
Дефицит GLUT1
Причина: SLC2A1 мутации
Проявления: Классический синдром: начало до 2 лет, редко старше.
Проявляется эпилептическими припадками, нарушением интеллектуального развития, дизартрией, микроцефалией, двигательными расстройствами (атаксия, дистония, хорея).
Атипичный вариант (в 10 % случаев): нет эпилепсии, пароксизмальные дискинезии — интермиттирующая атаксия, хореоатетоз, дистония, альтернирующая гемиплегия [16].
Аутосомно-рецессивные
Хорея-акантоцитоз
Причина: VPS13A мутации
Проявления: Двигательные расстройства: хорея, но возможен также паркинсонизм. Дистония с вовлечением оробукколингвальной области, как следствие — дизартрия, дисфагия и снижение массы тела из-за недоедания. Двигательные нарушения со временем прогрессируют.
Когнитивные и поведенческие изменения по типу лобного синдрома.
Эпилептические припадки в 50 % случаев, также заболевание может ими манифестировать.
Прогрессирующие дистальные миопатия и амиотрофия, отмечается повышение уровня КФК, ЛДГ, АСТ и АЛТ.
Глазодвигательные нарушения.
Акантоциты в крови.
Средний возраст дебюта заболевания — 30 лет, однако возможно начало в первую декаду жизни и после 70 лет [17].
Болезнь Вильсона
Причина: ATP7B мутации
Проявления: Мутации в ATP7B, кодирующем трансмембранную медь-транспортирующую АТФазу, приводят к аккумуляции меди в печени, головном мозге и других органах. Клинически данное заболевание чрезвычайно вариабельно, однако во всех случаях встречается поражение печени.
Неврологическая манифестация наблюдается среди 18–68 % больных.
Средний возраст дебюта — 20–30 лет (наименьший зафиксированный возраст — 6 лет, наибольший — 72 года).
Самые частые симптомы:
Тремор + дистония (фокальная, сегментарная, мультисегментарная или генерализованная) или паркинсонизм (брадикинезия, ригидность, тремор [в покое, постуральный, кинетический; различной амплитуды и частоты; уни- и билатерально; чаще вовлекаются дистальные части верхних конечностей]).
Дизартрия, постуральная неустойчивость, атаксия, сиалорея и дисфагия.
Реже — хорея, эпилептические припадки, «синдром беспокойных ног», нейропатия, расстройства сна, миоклонус, вовлечение пирамидных путей, глазодвигательные расстройства, расстройства вкуса.
Глаза: кольца Кайзера-Флейшера, катаракта.
Печень: бессимптомное повышение трансаминаз, острый гепатит, острая печеночная недостаточность, цирроз.
Эндокринная система: нарушение толерантности к глюкозе, гипопаратиреоз, нарушения роста и полового созревания, гинекомастия, нарушение менструального цикла, невынашивание беременности.
Сердце: аритмия, кардиомиопатия.
Почки: тубулярная дисфункция, нефролитиаз и нефрокальциноз, аминоацидурия, гиперкальциурия и гиперфосфатурия.
Опорно-двигательная система: остеопороз, хондрокальциноз, остеоартрит, артралгии.
Кожа и придатки: гиперпигментация голеней и стоп, «лазурная лунула», анетодермия, ксеродерма, черный акантоз, подкожные липомы, дерматомиозит.
Кровеносная система: острая гемолитическая анемия.
Диагностические мероприятия включают выявление характерного синдрома «глаза панды» и нейровизуализацию; оптическая когерентная томография, способная определить отложения меди на радужке; осмотр с помощью щелевой лампы; лабораторные анализы: церулоплазмин, суточная экскреция меди с мочой, уровень не связанной с церулоплазмином меди, концентрация меди в печени (таблица) [18].
Таблица. Особенности лабораторной диагностики болезни Вильсона.

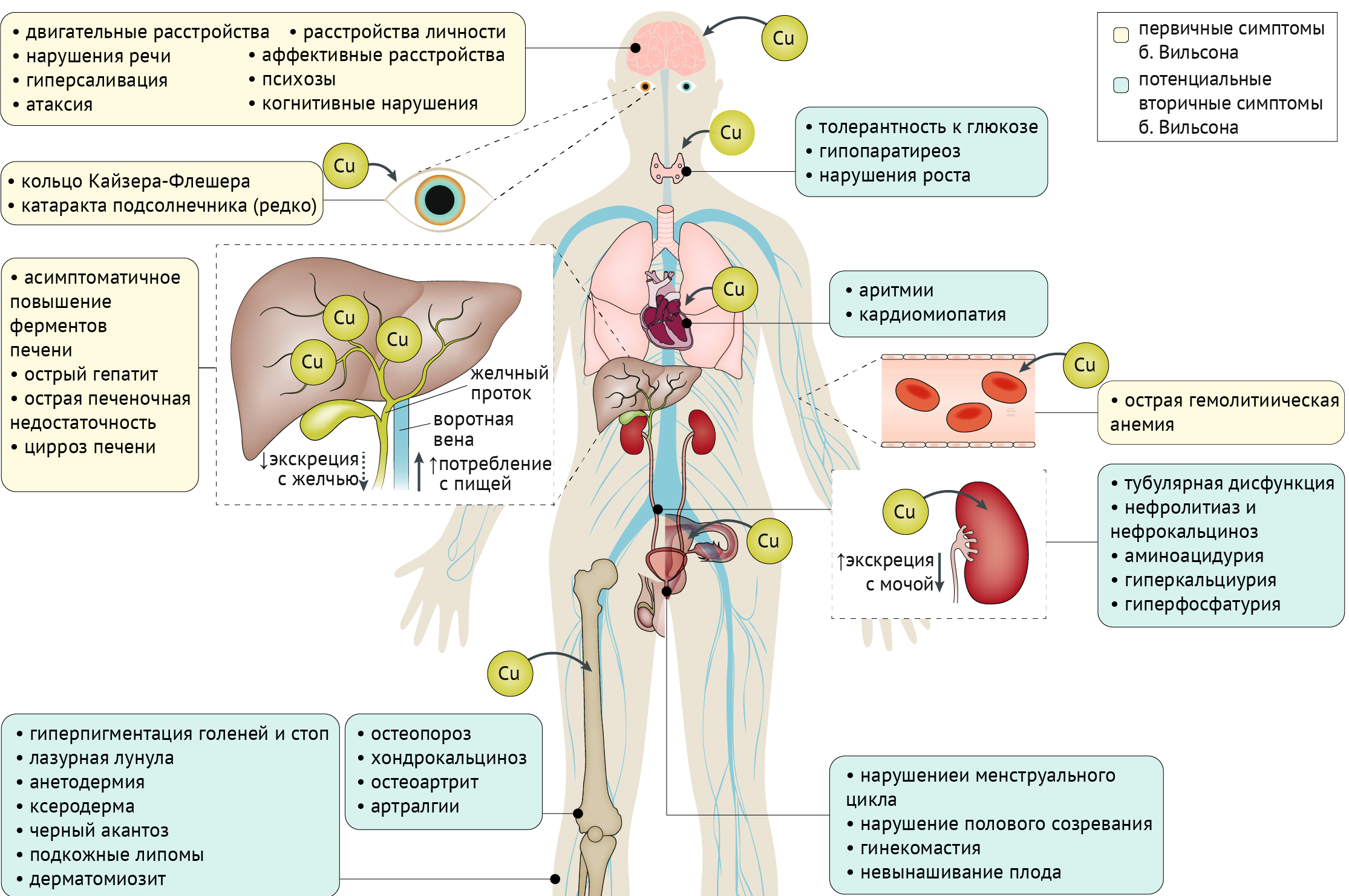
Рисунок 666. Симптомы болезни Вильсона
Хантингтон-подобное заболевание-3
Причина: Мало данных, возможная локализация мутации гена — 4p15.3
Заболевание описано у одной семьи, жителей Саудовской Аравии.
Проявления: Паркинсонизм, дистония, хорея, пирамидная симптоматика, атаксия, деменция.
Начало заболевания — 3–4 года [19].
Атаксия Фридрейха
Причина: Тринуклеотидные повторы GAA на 9q13-21.1 участке гена, кодирующего фратаксин; заболевание проявляется при наличии > 200 повторов
Проявления: Мозжечковая и сенсорная атаксия, аксональная сенсорная полинейропатия, мышечная слабость в дистальных отделах конечностей. Тремор лицевых мышц и верхних конечностей, хореоформные гиперкинезы, паркинсонизм. Нейропатия VII, X и XII черепных нервов, нарушение слуха и вертиго (вероятно, вследствие поражения VIII пары).
Деформации стоп, кифосколиоз.
Гипертрофическая кардиомиопатия и аритмии.
У 10 % — сахарный диабет.
Дебют заболевания — до 30 лет [20].
Атаксия-телеангиэктазия
Причина: Мутации гена ATM (ataxia-telangiectasia mutated)
Проявления: Атаксия выходит на первый план в первые 5 лет манифестации заболевания. Глазные и кожные телеангиэктазии появляются чуть позже, примерно к 3–6 годам. В последующем наблюдается прогрессирование атаксии, глазодвигательных нарушений и хореоатетоза (реже — дистонии), что приводит к инвалидизации и необходимости использования инвалидного кресла к 10–11 годам. Также возможны умеренные когнитивные расстройства.
Внешний вид пациента: «потерянный взгляд», седые волосы, глазные и кожные телеангиэктазии, сутулость, участки гипер- и гипопигментации кожи.
Повышен уровень α-фетопротеина.
Мутации гена ATM приводят к развитию иммунодефицита, частым инфекциям лицевых пазух и легких, стократному повышению риска онкологических заболеваний (85 % неоплазий — лимфомы или острая лимфоцитарная лейкемия).
Средняя продолжительность жизни — 20 лет. Основные причины смерти — бронхолегочные инфекции и онкологические заболевания [21].
Атаксия с окуломоторной апраксией 1 типа
Причина: мутации в APTX (aprataxin)
Проявления: Окуломоторная апраксия, атаксия, аксональная нейропатия, хорея и дистония верхних конечностей.
Низкий сывороточный альбумин, высокий уровень холестерина.
Средний возраст манифестации заболевания — 4,3 года (от 2 до 10 лет). К 15–25 годам появляется необходимость использования инвалидного кресла с целью передвижения [22].
Атаксия с окуломоторной апраксией 2 типа
Причина: мутации в SETX (senataxin)
Проявления: Мозжечковая атаксия, аксональная сенсомоторная нейропатия, окуломоторная апраксия, пирамидные симптомы, дистония рук, хореические гиперкинезы, тремор головы или постуральный тремор.
Дебют — от 3 до 30 лет.
Медленная прогрессия.
Повышенный уровень α-фетопротеина [23].
Нейродегенеративные заболевания, связанные с накоплением железа
Пантотенат киназа-ассоциированная нейродегенерация (PKAN)
Причина: PANK2 мутации
Проявления: Классический вариант: средний возраст манифестации — 3,4 года (6 мес–12 лет).
Неустойчивость, диспраксия, расстройства двигательного и когнитивного развития.
Спастичность нижних конечностей, дистония; ригидность, хореоатетоз, редко — паркинсонизм.
Пигментная ретинопатия в ⅔ случаев.
Атипичный вариант: средний возраст манифестации — 14 лет (1–28 лет).
Чаще проявляется нарушениями речи в виде палилалии или дизартрии, вокальных и моторных тиков. Треть пациентов имеют психиатрические расстройства. Дистония также свойственна атипичному варианту, хотя и менее выражена, нежели при классическом.
Характерная находка при PKAN на Т2 и FLAIR МРТ — «глаз тигра» [24].
Фосфолипаза A2G6-ассоциированная нейродегенерация (PLAN)
Причина: PLA2G6 мутации
Проявления: Классический вариант (инфантильная нейроаксиальная дистрофия): манифестация от 6 месяцев до 3 лет. Обычно до 6 месяцев болезнь протекает бессимптомно. Ранними проявлениями заболевания считаются регресс приобретенных навыков, неустойчивость, мышечную осевую гипотонию (проявляющуюся в неспособности поддерживать голову в определенном положении); в дальнейшем развиваются тетраплегия, страбизм и нистагм.
Атипичный вариант: развивается в период с раннего детства до конца второй декады жизни.
Атаксия, расстройства речи и социального взаимодействия, психические нарушения, дистония. В некоторых случаях с течением времени может развиться спастический тетрапарез.
PLA2G6-обусловленная дистония-паркинсонизм: манифестирует в период от 4 до 30 лет. Для раннего возраста наиболее характерна симптоматика, схожая с атипичным вариантом. Дистония-паркинсонизм + когнитивные и психиатрические нарушения свойственны лицам, начало заболевания для которых пришлось на поздний подростковый период. В более позднем возрасте на первый план выходит мозжечковая симптоматика или психиатрические расстройства.
Нейровизуализация при классическом варианте демонстрирует атрофию мозжечка, истончение и элонгацию валика мозолистого тела и характерный для этого варианта симптом гипертрофии тонкого бугорка (claval hypertrophy). При атипичном или PLA2G6 вариантах отмечаются атрофия мозжечка и аккумуляция железа в базальных ганглиях.
Ацерулоплазминемия
Причина: Мутации в CP
Проявления: Дебют заболевания — от 30 до 70 лет и позднее.
Характерна триада симптомов: дегенерация сетчатки, сахарный диабет, неврологическая симптоматика.
Неврологический аспект включает блефароспазм, дистонию лица и шеи, тремор, хорею, мозжечковую атаксию, когнитивные нарушения.
Кроме того, заболевание дебютирует с железодефицитной анемии.
Лабораторная диагностика включает в себя исследование уровней церулоплазмина, сывороточной меди, железа, ферритина, активности фермента церулоплазмин-ферроксидазы.
На МРТ наблюдаются отложения железа в печени, стриатуме, таламусе и мозжечке [25].
Другие педиатрические метаболические заболевания
Глутарическая ацидурия (ацидемия) первого типа (GA-1)
Причина: GCDH мутации
Проявления: Ранняя инфантильная форма (встречается наиболее часто): дебют заболевания приходится на возраст от 3 месяцев до 6 лет. При отсутствии раннего скрининга и своевременного начала терапии (что встречается среди 80–90 % детей с GA-1) развивается острая энцефалопатия (мышечная гипотония, потеря двигательных навыков, нарушение питания, припадки); с вероятностью 95 % это происходит в первые 24 месяца жизни. Энцефалопатия может быть вызвана интеркуррентным заболеванием, фебрильной реакцией на прививки, питание или стресс, связанный с анестезией и хирургическими вмешательствами; это приводит к острому двустороннему повреждению полосатого тела (как правило, в возрасте от 3 месяцев до 3 лет, в редких случаях от 3 до 6 лет) и сопровождается прогрессирующими комплексными неврологическими двигательными нарушениями (дистония, хорея и другие гиперкинезы). Вероятность инвалидизации и смерти кратно повышается после развития энцефалопатии.
Характерными для таких пациентов также являются макроцефалия, субдуральные кровоизлияния, эпилепсия и инфантильные спазмы.
Скрининг, диета и интенсивное лечение инфекционных и других заболеваний снижает риск развития острой энцефалопатии до 10–20 %. На МРТ: открытая оперкула, вентрикуломегалия, усиленный сигнал с базальных ганглиев, аномальный сигнал с белого вещества, субдуральные кровоизлияния, фронтотемпоральная гипоплазия.
Лабораторная диагностика: глутаровая кислота, 3-гидроксиглутаровая кислота, глутарил КоА, глутаминовая кислота.
Поздняя GA-1: манифестация после 6 лет. Симптоматика включает хронические головные боли, макроцефалию, эпилепсию, тремор и деменцию. Кроме того, были описаны периферическая нейропатия и возможные ассоциации с опухолями мозга. На МРТ: фронтотемпоральная гипоплазия и аномальный сигнал от белого вещества [26].
Пропионовая ацидемия (ПА)
Причина: Патогенные варианты PCCA или PCCB
Проявления: Неонатальная ПА (наиболее распространенная форма): в первые несколько дней жизни — сниженный аппетит и сонливость, далее развивается прогрессирующая энцефалопатия.
Если лечение не начато своевременно, энцефалопатия проявляется вялостью, судорогами и/или комой, что может привести к смерти. Состояние часто сопровождается метаболическим ацидозом с анионным дисбалансом, лактатацидозом, кетонурией, гипогликемией, гипераммониемией и панцитопенией.
ПА с поздним началом: может протекать как бессимптомно с периодами метаболических нарушений в условиях катаболического стресса (например, болезни, операции, голодания), так и демонстрировать начало заболевания с развития полиорганной недостаточности, включая рвоту, непереносимость белка, задержку развития или даже его регресс, мышечную гипотонию, двигательные расстройства или кардиомиопатию.
ПА в редких случаях может проявляться только изолированной кардиомиопатией при отсутствии клинической метаболической декомпенсации или нейрокогнитивных нарушений.
По мере развития заболевания проявления ПА могут включать нарушения роста, умственную отсталость, судороги, поражения базальных ганглиев, панкреатит и кардиомиопатию. Другие редко встречающиеся осложнения включают атрофию зрительного нерва, потерю слуха, преждевременную недостаточность яичников и хроническую почечную недостаточность.
Скрининг включает обследование новорожденных на 3-гидроксипропионат, метилцитрат, тиглиглицин, пропионилглицин и молочную кислоту [27].
X-сцепленные заболевания
Синдром Мак-Лауда
Причина: XK мутации
Относится к нейроакантоцитозам, для которых характерно присутствие акантоцитов в крови.
Проявления со стороны ЦНС: синдром прогрессирующей хореи, сходный с синдромом Хантингтона, включающий клиническую триаду: двигательные расстройства, когнитивные нарушения и психиатрические проявления; эпилептические судороги, в основном генерализованные.
Нейромышечные проявления (часто субклинические или легкие): сенсомоторная аксонопатия; нейрогенная мышечная атрофия, включая необъяснимое повышение креатинфосфокиназы; миопатия; кардиомиопатия.
Нарушения ритма сердца: тахиаритмии и фибрилляция предсердий.
Вышеописанные проявления наиболее характерны для мужчин (учитывая X-рецессивное наследование).
Среди женщин возможно развитие хореиформных гиперкинезов, тиков и/или сенильной деменции.
Манифестация заболевания приходится на период от 7 до 51 года. Пациенты погибают к 53-летнему возрасту (в среднем от 31 до 69 лет) [28].
Синдром Леши-Нихена
Причина: HPRT1 мутации
Проявления: Заболевание обусловлено недостаточностью фермента гипоксантин-гуанинфосфорибозилтрансферазы, в результате чего образуется большое количество мочевой кислоты.
Синдром характеризуется двигательной дисфункцией, напоминающей церебральный паралич, когнитивными и поведенческими нарушениями и гиперурикемией.
Наиболее часто встречающиеся признаки (гипотония и задержка развития) проявляются в возрасте от 3 до 6 месяцев.
В течение первых нескольких лет развиваются экстрапирамидные (дистония, хореоатетоз, опистотонус) и пирамидные нарушения (спастичность, гиперрефлексия, разгибательные подошвенные рефлексы).
Когнитивные и поведенческие расстройства проявляются в возрасте от 2 до 3 лет. Отличительной чертой этого заболевания является характерная самотравматизация.
Гиперурикемия может привести к отложению кристаллов или камней мочевой кислоты в почках, мочеточниках или мочевом пузыре, позже развивается подагрический артрит.
Более мягкие формы заболевания с менее выраженными проявлениями включают гиперурикемию с неврологическими расстройствами, но без самотравматизации; только гиперурикемию, иногда с острой почечной недостаточностью.
Хотя женщины-носители патогенной хромосомы, как правило, не имеют симптомов заболевания, у них может быть обнаружена гиперурикурия, симптомы которой могут развиться и в более поздние годы [29].
Синдром Любага (X-связанная дистония-паркинсонизм)
Причина: XDP мутации
Проявления: Заболевание в первую очередь характерно для филиппинцев. Дебют приходится в среднем на 39 лет.
Дистония различной степени тяжести, от очаговой до генерализованной, обычно манифестирующая в раннем взрослом возрасте; паркинсонизм (в некоторых случаях отвечающий на прием леводопы).
Двигательный (акционный) тремор или тремор покоя, хорея, миоклонус.
Когнитивные расстройства встречаются редко, проявляются в нарушениях лобной исполнительной функции. У пациентов также может наблюдаться депрессия.
Обонятельные нарушения сильнее выражены у пациентов с синдромом Любага, нежели при болезни Паркинсона.
Худший прогноз наблюдается при развитии паркинсонизма и дистонии оробукколингвальной области и шеи в течение года или двух после манифестации заболевания. Болезнь приковывает к постели и приводит к смерти уже в течение 2–5 лет после дебюта, к наиболее распространенным причинам смерти относятся аспирационная пневмония, гортанный стридор и/или интеркуррентные инфекции, возникающие в результате иммобилизации.
У женщин-носителей болезнь, как правило, протекает бессимптомно, однако по мере развития заболевания возможна манифестация схожей симптоматики, хотя и куда менее выраженной, чем у мужчин.
КТ/МРТ обычно не дает ценной информации. Диагностика включает выявление мутации и ПЭТ [30].
Митохондриальные заболевания
Синдром Лея (Ли)
Причина: Мутации MT-ATP6, MT-ND3, MT-ND5, MT-ND6, MT-CO3, MT-ND1, MT-ND2, MT-ND4, MT-TI, MT-TK, MT-TL1, MT-TL2, MT-TV, MT-TW.
Проявления: Синдром Ли (подострая некротическая энцефаломиелопатия) характеризуется проявлением симптомов в возрасте от 3 до 12 месяцев, часто после вирусной инфекции. Декомпенсация (нередко сопровождающаяся повышенным уровнем лактата в крови и/или цереброспинальной жидкости [ЦСЖ]) во время интеркуррентного заболевания обычно ассоциируется с психомоторной отсталостью или регрессом. Неврологические особенности включают гипотонию, спастичность, двигательные расстройства (в т. ч. хорею), мозжечковую атаксию и периферическую невропатию, эпилептические припадки, нистагм, офтальмопарез, атрофию зрительного нерва. Иногда развивается гипертрофическая кардиомиопатия. 50 % пациентов умирают в возрасте до трех лет, чаще всего в результате дыхательной или сердечной недостаточности.
NARP — проксимальная нейрогенная мышечная слабость с сенсорной нейропатией, атаксией и пигментной ретинопатией — заболевание, характеризующееся атаксией и трудностями в обучении, часто возникающими в раннем детстве. Состояние больных NARP остается относительно стабильным в течение многих лет, но могут также наблюдаться эпизодические ухудшения, часто в связи с вирусными заболеваниями. В дальнейшем к симптоматике также присоединяется эпилепсия [31].
Причина: Мутации MT-TL1, MT-ND5, MT-TC, MT-TF, MT-TH, MT-TK, MT-TL2, MT-TQ, MT-TV, MT-TW, MT-TS1, MT-TS2, MT-ND1, MT-ND6, MT-CO2, MT-CO3, MT-CYB.
Митохондриальная энцефалопатия, лактацидоз и инсультоподобные эпизоды.
Дебют заболевания наблюдается в среднем между 2 и 40 годами (5–8 % пациентов заболевают в возрасте младше 2 лет; 1–6 % — старше 40 лет).
Проявления: инсультоподобные эпизоды в возрасте до 40 лет; энцефалопатия с судорогами и/или деменцией; периодические головные боли; мышечная слабость и непереносимость физических нагрузок; корковая слепота; гемипарез; периодическая рвота; невысокий рост; нарушение слуха; нормальное раннее психомоторное развитие; периферическая нейропатия; трудности в обучении.
Лактацидоз, повышение уровня лактата в крови и ЦСЖ.
На МРТ головного мозга во время инсультоподобных эпизодов наблюдается усиление Т2-сигнала, не соответствующее классическому распределению сосудов (отсюда и термин «инсультоподобный»). Также отмечаются асимметричность анатомических структур головного мозга; обычно вовлекаются преимущественно височные, теменные и затылочные доли;
Патологический процесс ограничен корой или вовлекает субкортикальное белое вещество.
Медленное распространение инсультоподобных поражений на МРТ происходит в течение нескольких недель после появления первых симптомов.
Диффузионно-взвешенная МРТ демонстрирует повышенный коэффициент диффузии (КД) в зонах инсультоподобных поражений, в отличие от ишемических инсультов, при которых наблюдается снижение КД.
МР-ангиография для диагностики не информативна, а МР-спектроскопия показывает снижение сигнала N-ацетиласпартата и накопление лактата [32].
Приобретенные/спорадические заболевания
Индуцированные препаратами:
Тардивная дискинезия, побочные эффекты приема препаратов.
Иммуно-индуцированные:
Хорея Сиденгама
Аутоиммунные:
СКВ, АФС, другие СЗСТ/васкулиты ЦНС, паранеопластические синдромы
Инфекции:
ВИЧ/СПИД ассоциированные: ВИЧ-энцефалопатия, токсоплазмоз, лимфома;
Другие нейроинфекции:
Вирусный энцефалит, микоплазменный энцефалит, цистицеркоз, нейросифилис.
Неоплазии:
Первичная или метастатическая опухоль мозга, лимфома ЦНС.
Эндокринопатия:
Гипертиреоидизм, гипо/гиперпаратиреоидизм, хорея беременных.
Метаболические:
Гипер/гипогликемия, дисбаланс электролитов, приобретенная гепатоцеребральная дегенерация, дефицит B12.
Сосудистые:
Субкортикальный/базальноганглионарный инсульт, сосудистые мальформации, болезнь Моя-Моя.
Спорадические нейродегенеративные заболевания:
Болезнь Альцгеймера, кортико-базальная дегенерация, болезнь Крейтцфельда-Якоба в рамках спорадической прионопатии.
Другие:
Истинная полицитемия, Postpump-хорея, рассеянный склероз, токсины, гипоксическая-ишемическая энцефалопатия.
Для поиска причины заболеваний, одним из симптомов которых является хорея (примеры заболеваний представлены в табл. 2), необходимо учитывать течение заболевания, наследственность, прием определенных препаратов, а также данные нейровизуализации и лабораторных анализов. Алгоритм диагностики показан ниже (схема 7) [33]:
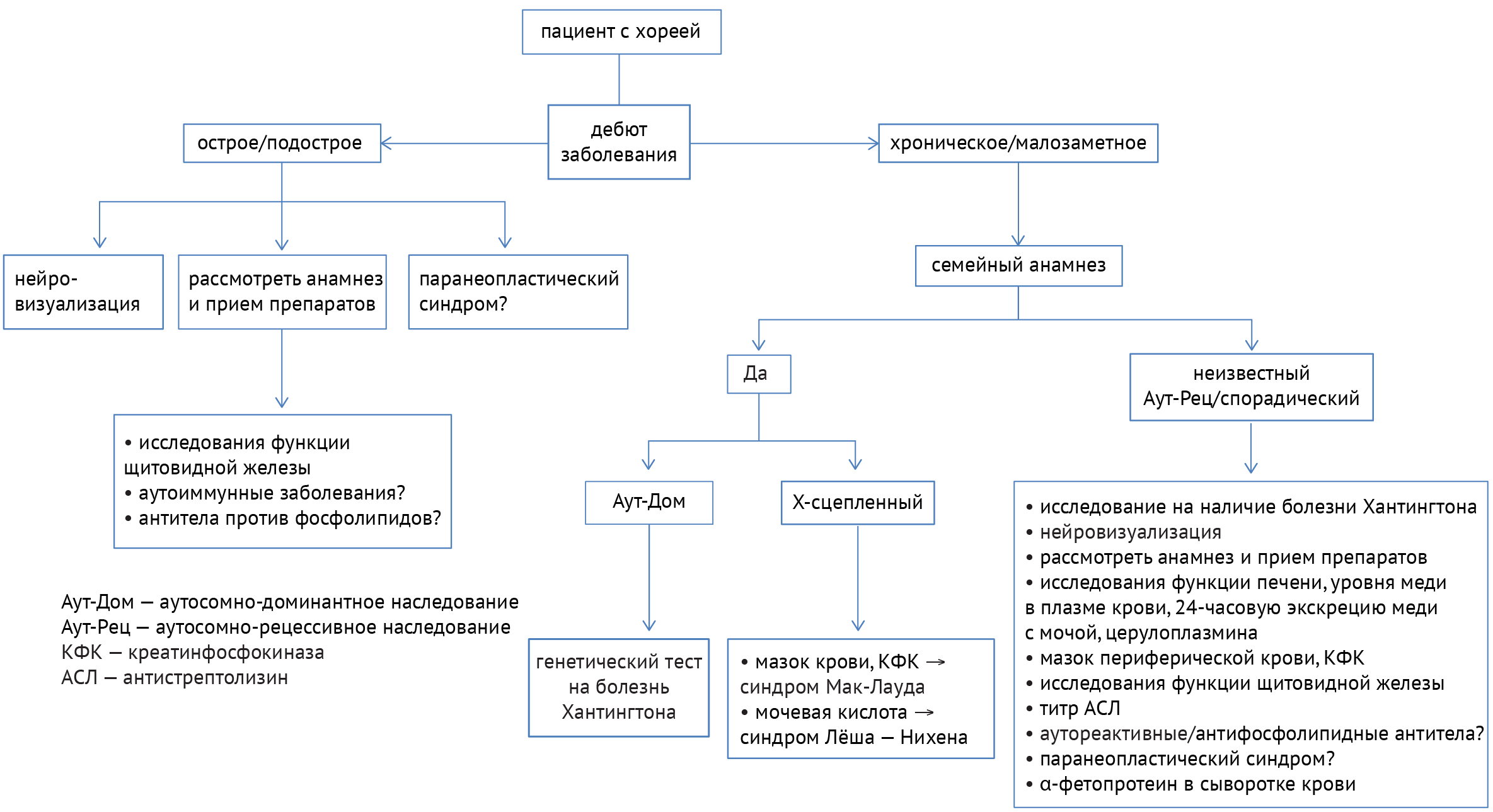
В дифференциальной диагностике заболеваний с симптомами хореи требуется разграничивать наследственные и приобретенные расстройства. В случае установленного наследственного заболевания следует пользоваться следующим алгоритмом (схема 8):
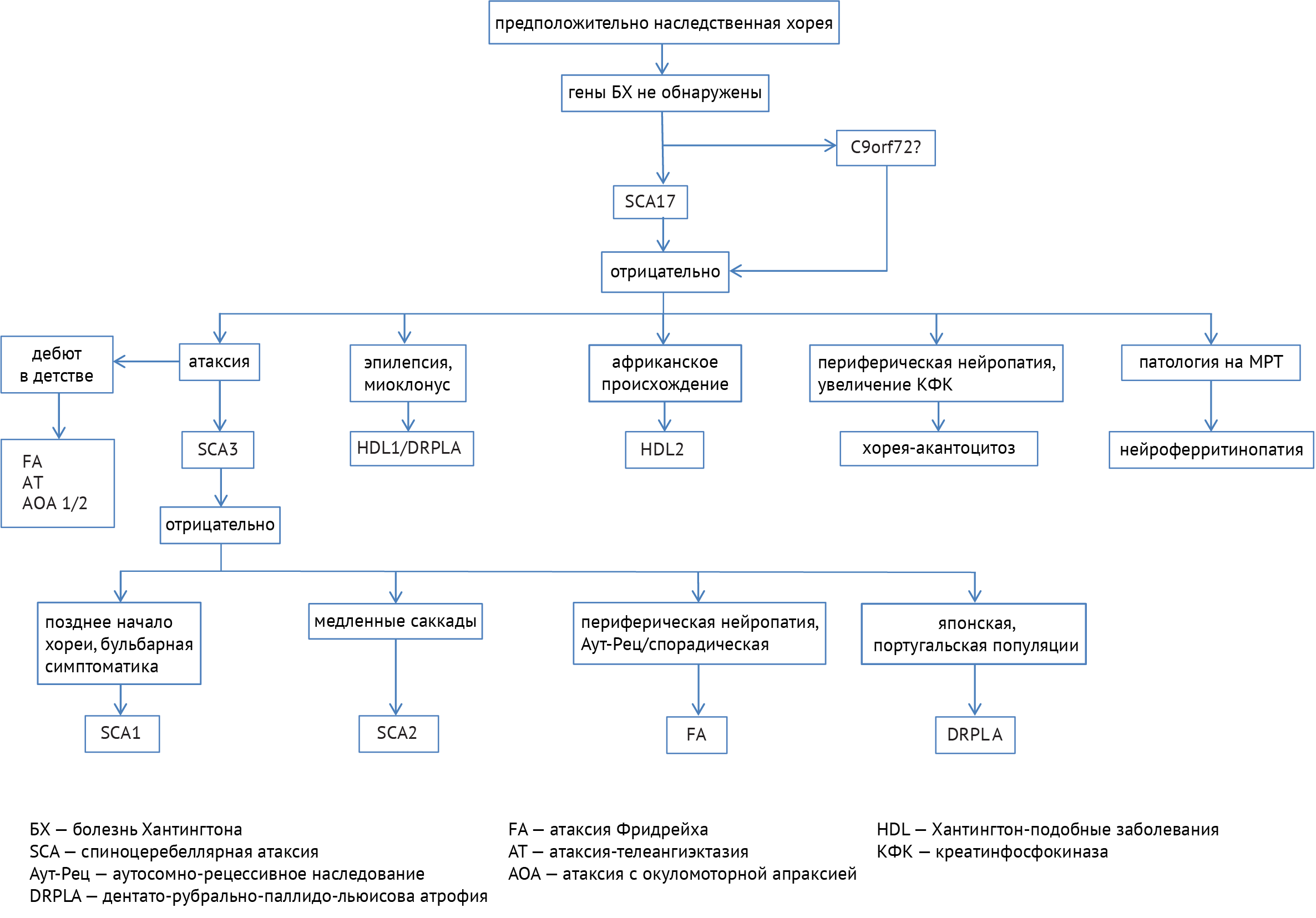
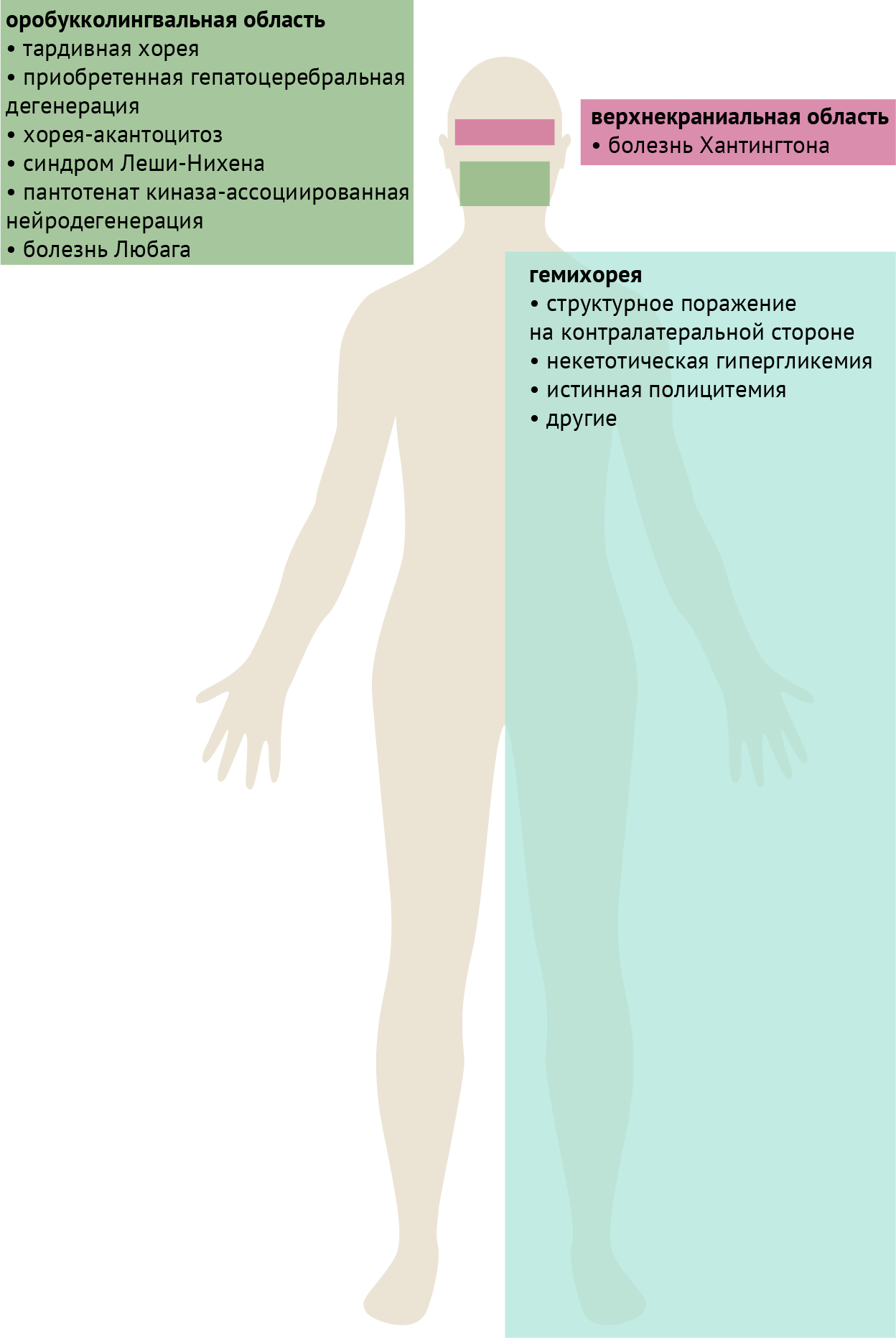
Также для некоторых заболеваний можно выделить характерные топографические особенности, которые, тем не менее, не являются абсолютно специфическими и/или чувствительными признаками (рис. 5).
Гемихорея. Поражения, приводящие к гемихорее, классически локализуются в контралатеральном субталамическом ядре. Тем не менее, они не ограничиваются этой структурой и могут располагаться в других частях контралатеральных базальных ганглиев или в лучистой короне. Системные расстройства, такие как некетотическая гипергликемия и истинная полицитемия, также могут проявляться гемихореей, либо иметь совершенно асимметричный характер поражения. Хорея Сиденгама также может быть асимметричной, проявляться гемихореей или даже принимать вид гемибаллизма. К структурным поражениям также относятся описанные в табл. 2 сосудистые заболевания и неоплазии.
Оробукколингвальная область. Хореические движения в оробукколингвальной или нижней краниальной областях классически проявляются при тардивном синдроме и приобретенной гепатоцеребральной дегенерации. Следует отметить, что более академичным феноменологическим термином в данном случае является «стереотипии при тардивном синдроме с вовлечением оробукколингвальной области», поскольку движения имеют часто повторяющийся паттерн. При тардивном синдроме в оробукколингвальной области могут наблюдаться латеральные движения языка, как будто у пациента за щекой леденец. Дискинезия, вызванная леводопой, при мультисистемной атрофии имеет тенденцию вовлекать ту же область, в отличие от дискинезии конечностей при классической болезни Паркинсона.
Нейроакантоцитоз представляет собой группу расстройств, при которых прогрессирующая нейродегенерация связана с наличием в крови акантоцитов. Двумя основными заболеваниями этой группы являются хорея-акантоцитоз (аутосомно-рецессивное наследование) и синдром Мак-Лауда (Х-сцепленное наследование). Хорея-акантоцитоз может протекать с выраженной дистонией языка, особенно во время принятия пищи или при кормлении, что может вызывать травматизацию языка зубами. Напротив, при синдроме Мак-Лауда вовлечение оробукколингвальной области, включая дистонию при кормлении, встречается гораздо реже. Следует отметить, что при дифференциальной диагностике расстройств, которые могут сопровождаться оромандибулярной дистонией и самотравматизацией, также следует рассмотреть синдром Леша-Нихена. Это заболевание, передающееся по X-рецессивной хромосоме, вызванное дефектом фермента гипоксантин-гуанинфосфорибозилтрансферазы и проявляющееся гиперурикемией, которая обычно развивается в течение первых нескольких лет жизни, тогда как дебют хореи-акантоцитоза обычно наступает в раннем взрослом возрасте.
Хорея верхней краниальной области. При данном заболевании наблюдаются хореиформные движение бровей и мышц лба. Для тардивной дискинезии обычно не характерно вовлечение этой области, в отличие от хореи Хантингтона, однако возможны и обратные случаи.
Лечение хореи состоит из этиологического и симптоматического этапов.
Симптоматическая терапия
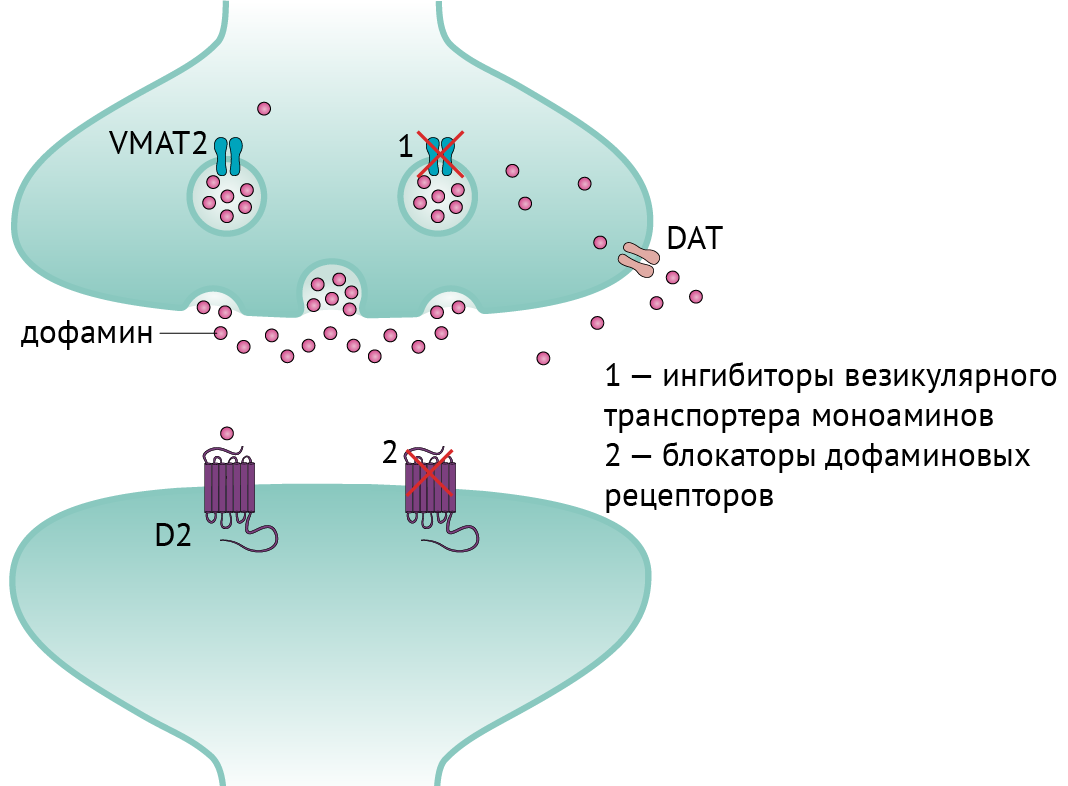
◇ Препараты, снижающие уровень пресинаптического дофамина (рис. 6):
→ ингибиторы везикулярного моноаминового транспортера (VMAT): тетрабеназин и препараты нового поколения — деутетрабеназин, валбеназин (ингибиторы VMAT-2) и резерпин (ингибитор VMAT-1). Ингибиторы VMAT-1 действуют на периферическую нервную систему, что может привести к гипотензии. Основные побочные эффекты ингибиторов VMAT — паркинсонизм, депрессия и акатизия. Кроме того, деградация препарата осуществляется ферментом CYP2D6, так что, например, при превышении дозировки тетрабеназина более 50 мг/сут следует отслеживать уровень CYP2D6.
◇ Блокаторы постсинаптических дофаминовых рецепторов:
→ типичные антипсихотики (например, галоперидол);
→ атипичные антипсихотики (например, оланзапин, рисперидон, кветиапин, клозапин).
◇ Другие:
→ антиконвульсанты (например, вальпроевая кислота, карбамазепин).
Глубокая стимуляция мозга:
◆ Подтверждена эффективность данного метода при болезни Хантингтона, хорее-акантоцитозе, приобретенной хорее.
Основы фармакологического вмешательства — «начинай с малого, продвигайся медленно». Терапия начинается с минимально допустимой дозировки; проводится постепенное титрование дозы, целью которого является максимально возможное снижение побочных явлений от препаратов.
Миоклонии
Миоклонии — быстрые, молниеносные, внезапные, не волевые подергивания/вздрагивания одной мышцы или групп мышц [34].
В отличие от фасцикуляций, где подергивания проявляются в единичных мышечных волокнах и пучках, сокращения мышцы и группы мышц при миоклонусе могут привести к нарушению устойчивости, падениям и нарушению качества жизни.
В первую очередь при диагностике вынужденных подергиваний мышц следует уметь определять вероятную причину этого состояния (табл. 3).
Таблица 3. Дифференциальная диагностика миоклоний

Классификация миоклонуса по наличию триггера:
Миоклонус может индуцироваться как мышечной активностью (позитивный), так и ее внезапным прекращением (негативный). Описаны три типа негативного миоклонуса: астериксис (трепетание рук при разгибании запястья) у пациентов с токсико-метаболической энцефалопатией; негативный миоклонус, вовлекающий аксиальную мускулатуру и нижние конечности, приводящий к шаткой походке и спонтанным падениям; эпилептический негативный миоклонус. Последний определяется как прерывание мышечной активности, привязанное к эпилептической аномалии ЭЭГ при условии отсутствия ранее возникшего позитивного миоклонуса. Эпилептический негативный миоклонус может наблюдаться при множестве эпилептических расстройств; негативный и позитивный миоклонусы могут сочетаться, например, среди прогрессивных эпилептических миоклоний.
Миоклонус может быть индуцированным (активируется в результате соматосенсорной провокации [тактильной, зрительной, слуховой]) и спонтанным (миоклонии без триггера).
Анатомическая локализация субстрата миоклонуса представлена в табл. 4. Конечно, стоит помнить, что возможны разнообразные сочетания, например, кортикальной и субкортикальной миоклоний.
Таблица 4. Анатомический субстрат миоклоний.
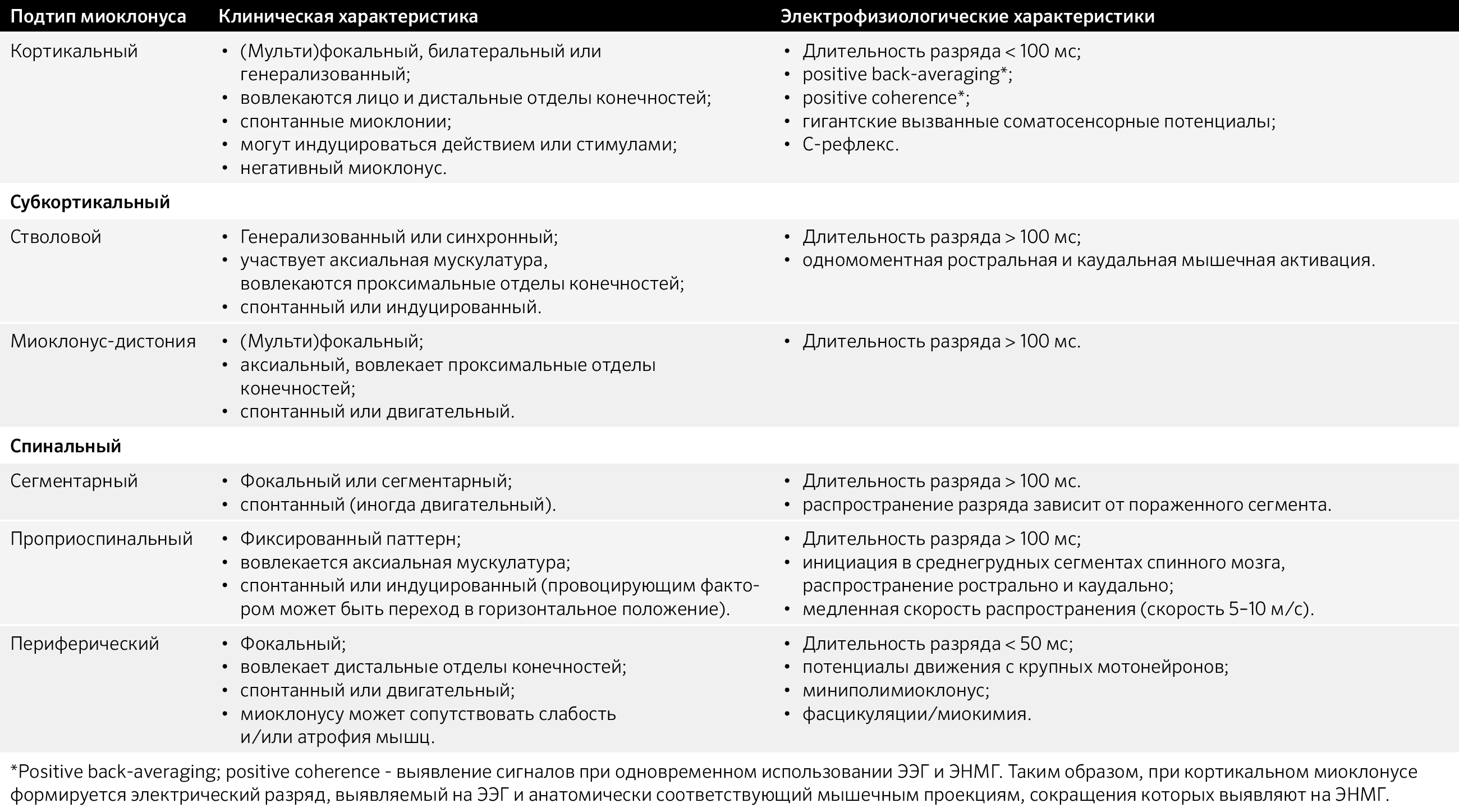
Субкортикальный миоклонус может быть сегментарным и несегментарным. Несегментарный в свою очередь подразделяется на физиологический (стартл-рефлекс) и патологический (гиперэкплексия — избыточный стартл-рефлекс; ретикулярный рефлекс).
Стартл-рефлекс является физиологической реакцией на внезапные стимулы (например, слуховые), что в свою очередь сопровождается вздрагиванием «мантийной зоны» (головы, лица, верхнего плечевого пояса). Грубо говоря, если человека напугать, он дернется — это и есть стартл-синдром. Активность (согласно данным ЭНМГ) начинается в грудино-ключично-сосцевидной мышце и распространяется на лицо, туловище и конечности. Гиперэкплексия — это избыточный стартл-рефлекс. Даже если пациент ожидает стимула, гиперэкплексия всё равно приводит к генерализованному миоклонусу. Она может быть семейной (табл. 7), идиопатической или возникать в результате демиелинизирующих заболеваний, сосудистых поражений и стволового энцефалита [33].
Ретикулярный рефлекс отличается от гиперэкплексии тем, что первый с гораздо большей вероятностью может оказаться спонтанным, а также активироваться через соматосенсорную стимуляцию дистальных отделов конечностей, нежели чем «мантийной зоны». Ретикулярный рефлекс может являться следствием постгипоксической энцефалопатии, стволового энцефалита и уремии.
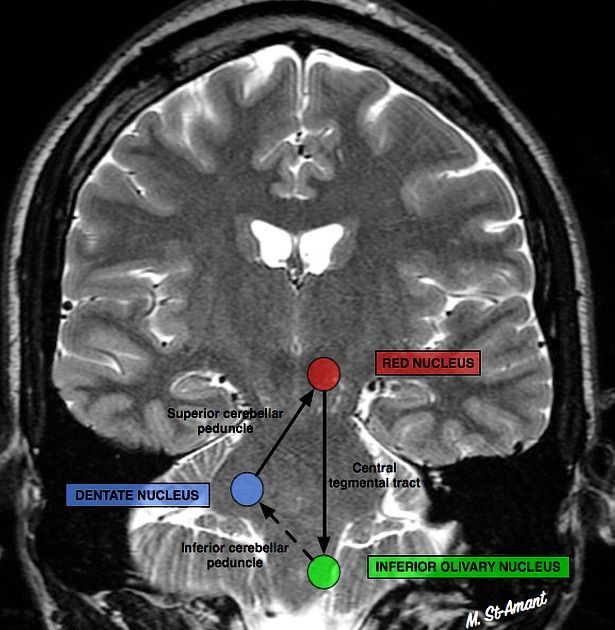
Сегментарный миоклонус: эссенциальный и симптоматический миоклонус неба, иногда рассматривающийся как тремор. Эссенциальный обусловлен дисфункцией, а симптоматический — структурным поражением области треугольника Молляре (в проекции которого локализуются связи между зубчатым ядром мозжечка, красным ядром и нижним оливарным ядром) (рис. 7) [36].
Эссенциальный миоклонус неба (ЭМН) (в 1990 году был переименован в тремор, однако применение обоих терминов не является ошибочным — прим. авт.) проявляется ритмичными сокращениями мышцы, напрягающей небную занавеску, отходящей от боковой стенки евстахиевой трубы. Открытие и закрытие трубы в результате мышечного сокращения производит слышимый «щелчок». ЭМН исчезает при засыпании. При симптоматическом миоклонусе вовлекается мышца, приподнимающая небную занавеску, и он, в отличие от ЭМН, не сопровождается «щелчком», а также не исчезает во время сна. Симптоматический миоклонус чаще всего проявляется при сосудистых, онкологических и демиелинизирующих заболеваниях ствола мозга. Другая хорошо известная его причина — синдром прогрессивной атаксии и тремора неба (PAPT — progressive ataxia palatal tremor syndrome).
Что касается спинального миоклонуса, стоит обратить внимание на то, что проприоспинальный миоклонус можно спутать со стволовым генерализованным миоклонусом, так как и в том, и в другом случаях вовлекается аксиальная мускулатура. Однако проприоспинальный миоклонус не распространяется на лицевую мускулатуру и не провоцируется слуховыми стимулами.
Разные типы миоклонуса имеют разную этиологию и, следовательно, требуют разных клинических подходов. Корковый и подкорковый типы миоклоний могут быть как приобретенными, так и наследственными, в отличие от спинального и периферического миоклонусов, которые обычно являются приобретенными.
Этиологическая классификация миоклоний
Физиологический миоклонус — возникает у здоровых людей (гипнические подергивания; подергивания в результате физической нагрузки, повышенной тревожности; икота).
Эссенциальный миоклонус — как правило, генетически детерминированный (спорадический или наследственный). Протекает изолированно либо выходит на первый план среди других симптомов заболевания. Обычно выраженного нарушения качества и продолжительности жизни не происходит.
Эпилептический миоклонус — возникающий при эпилепсии. Может быть как позитивным, так и негативным. Эпилептический миоклонус сопровождается эпилептиформными разрядами на ЭЭГ; по своим проявлениям может быть очаговым, сегментарным или генерализованным.
Если анатомический локус миоклоний определен как периферический или спинальный, необходимо оценить признаки денервации (клинический осмотр и/или ЭНМГ). Структурные поражения должны исследоваться с помощью соответствующего метода визуализации, дабы сузить возможные причины миоклонуса. Например, периферический миоклонус обычно возникает в результате повреждения периферической нервной системы — обычно это поражения плечевого сплетения или корешков спинного мозга, которые можно выявить с помощью ЭНМГ и иногда с помощью МРТ.
Повреждение спинного мозга может вызвать спинальные миоклонии.
Сегментарный миоклонус очень редок и почти всегда обусловлен структурными повреждениями спинного мозга. Острое или подострое начало, быстрое прогрессирование, радикулопатия или полирадикулопатия и системные признаки (лихорадка, кожная сыпь или поражение суставов) предполагают наличие инфекционной или аутоиммунной этиологии, которая должна быть подтверждена соответствующими лабораторными исследованиями. Важно отметить, что подавляющее большинство случаев проприоспинального миоклонуса в настоящее время считается функциональным расстройством движения. Кроме того, в редких случаях спинальный миоклонус может быть вызван лекарственными препаратами или инфекциями, что подчеркивает необходимость тщательного обследования пациентов с этим типом миоклонуса.
Корковый и подкорковый типы миоклоний должны быть отдифференцированы от широкого диапазона состояний. В целом, острое или подострое начало и/или быстрое прогрессирование миоклоний являются важными факторами для принятия решения в пользу приобретенной причины, тогда как заболевание с ранним началом и медленным прогрессированием более характерно для генетического расстройства. Специфические клинические признаки, которые сосуществуют с миоклонусом, часто предоставляют важную информацию относительно основного заболевания.
Схема, представленная ниже (схема 9), поможет разобраться в диагностических мероприятиях, направленных на выявление причины миоклонуса.
Схема 9. Восьмишаговый алгоритм диагностики миоклоний.
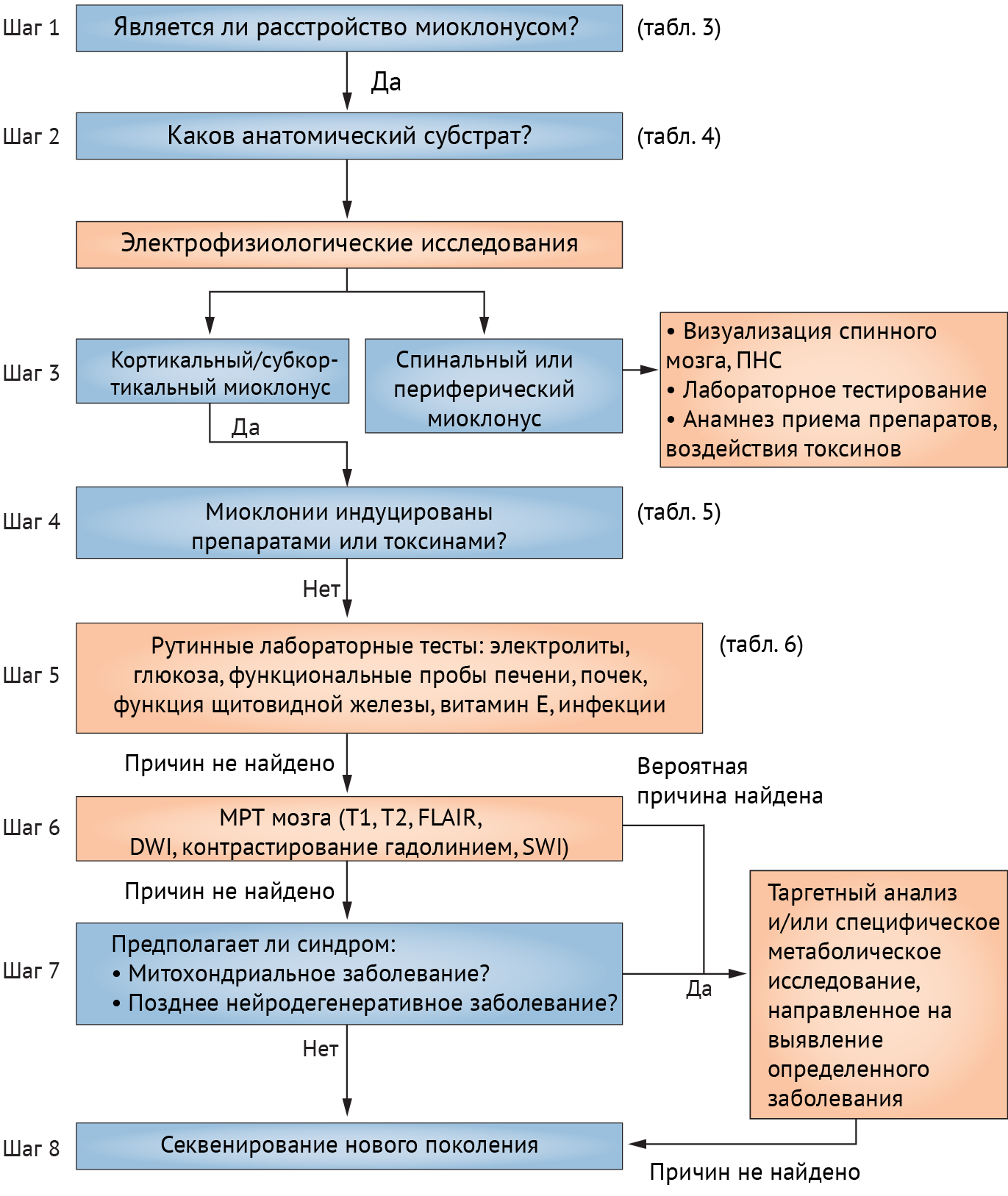
Таблица 5. Препараты и токсины, способные вызвать миоклонус.

Таблица 6. Приобретенные причины миоклоний.
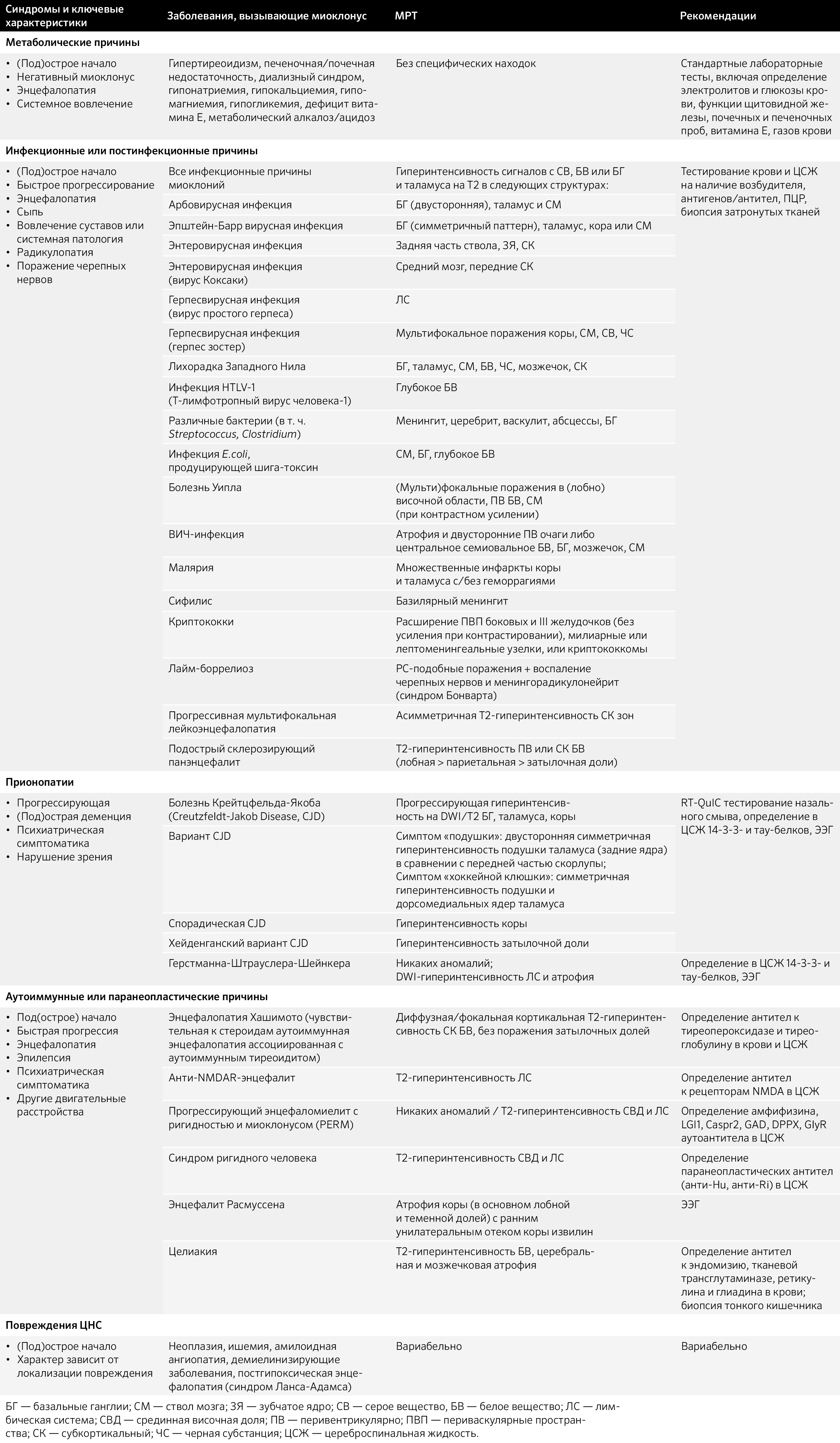
Таблица 7. Наследственные причины миоклонуса [37].
Таблица 7.1. Гиперэкплексия-синдром

7.2 Миоклонус-эпилепсия
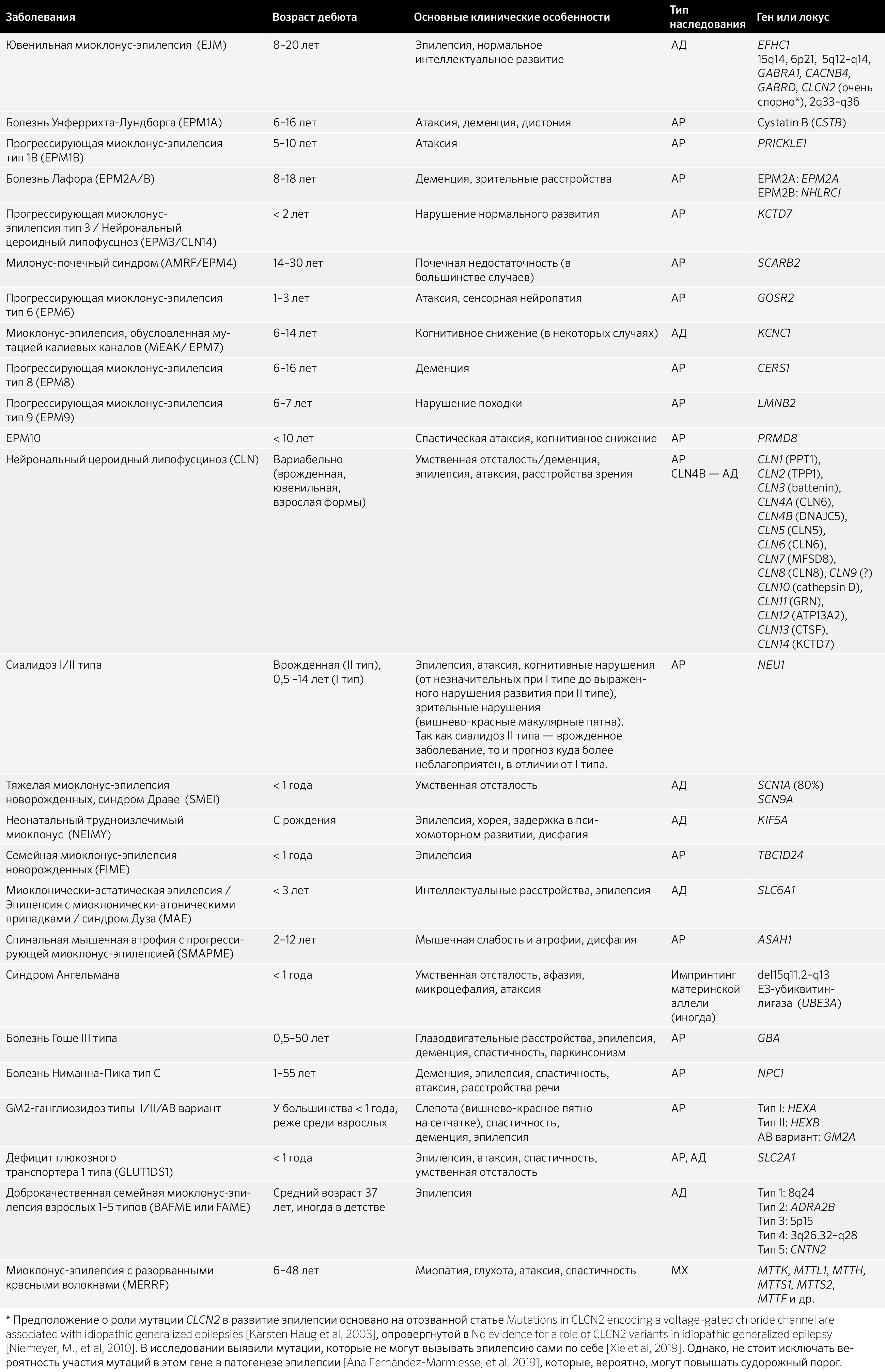
7.3 Нарушения развития нервной системы, двигательные расстройства и нейродегенеративные заболевания с миоклонусом
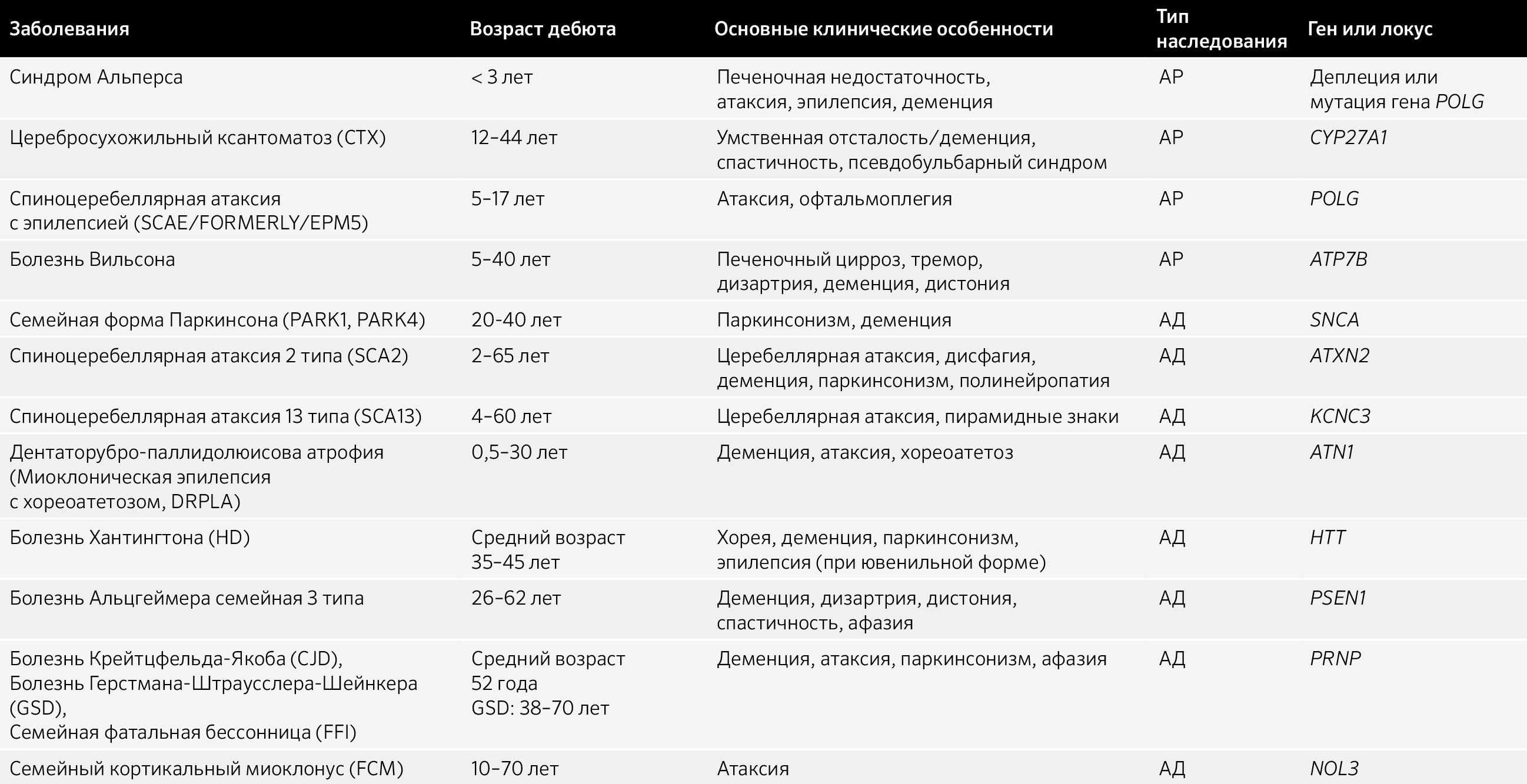
Таблица 7.4 Миоклонус-дистония

Терапия миоклонуса
Лечение миоклоний, в первую очередь, состоит в своевременной диагностике потенциально обратимых заболеваний и состояний, с целью оказания качественной помощи. Когда этиологическое лечение не представляется возможным или нужно подключить симптоматическую терапию — сперва необходимо определить тип миоклонуса по анатомической классификации (табл. 4.), т. к. при разных типах — эффективны различные препараты.
Для лечения миоклонуса зачастую требуется контролирование концентрации препаратов в крови с последующим повышением их дозировок, и кроме того, симптоматику редко удается контролировать одним единственным средством. В целом, противоэпилептические препараты (такие как вальпроат натрия, леветирацетам и пирацетам) эффективны при кортикальном миоклонусе, но неэффективны при других формах. Клоназепам может быть полезен при всех типах миоклоний.
Корковый миоклонус. Лечение кортикального миоклонуса направлено на усиление ГАМК-ергической нейротрансмиссии. Как правило, корковый миоклонус лечится комбинацией препаратов. Седация и атаксия являются основными побочными эффектами политерапии, но их можно преодолеть с помощью принципа «начинай с малого, иди медленно».
Среди ГАМК-ергических препаратов особенно эффективен вальпроат натрия. Препарат вводят медленно и титруют до 1200–2000 мг в день. Бензодиазепины также очень полезны, в частности клоназепам в больших дозах (до 15 мг в день). Однако толерантность к нему может развиться через несколько месяцев, в то время как быстрое снижение дозы или прекращение приема препарата может привести к заметному ухудшению.
Пирацетам и леветирацетам являются двумя родственными препаратами, эффективными при миоклонусе, хотя их точный механизм действия недостаточно изучен. Для терапии миоклоний могут потребоваться большие дозы пирацетама (3200–4800 мг в день, максимум до 20 г в день), но меньшие — леветирацетама (максимум 3000 мг в день). При кортикальном миоклонусе эти препараты активно используются как в качестве монотерапии, так и в сочетании с вальпроатом натрия и клоназепамом.
Примидон и фенобарбитал редко эффективны, в то время как зонисамид может быть полезен в некоторых случаях прогрессирующей миоклонус-эпилепсии.
Лучше всего избегать использования фенитоина, карбамазепина, ламотриджина и вигабатрина при кортикальном миоклонусе, т. к. они могут парадоксально усугублять симптоматику. Это особенно относится к использованию фенитоина при болезни Унферрихта-Лундборга. Лечение прогрессирующей миоклонус-эпилепсии является очень сложным, т. к. препараты, которые помогают при генерализованных припадках, могут ухудшать течение миоклоний, и наоборот.
Негативный миоклонус. Эпилептический негативный миоклонус (ЭНМ) у детей, страдающих идиопатической фокальной эпилепсией, может реагировать на этосуксимид и леветирацетам. ЭНМ, ассоциированный с структурной эпилепсией, обычно менее чувствителен к обычным противоэпилептическим препаратам и может усугубляться карбамазепином, вальпроевой кислотой, фенитоином, ламотриджином и окскарбазепином. Постгипоксический миоклонус, выраженный в дистальных отделах конечностей, и рефлекторный миоклонус верхних конечностей реагируют на терапию намного лучше, чем негативный миоклонус проксимальных мышц нижних конечностей, вызывающий нарушения походки и частые падения.
Подкорковый миоклонус. Противоэпилептические препараты, используемые при кортикальном миоклонусе, не эффективны при субкортикальном. Клоназепам полезен при гиперэкплексии и частично при ретикулярном рефлекторном миоклонусе. Миоклонус-дистония частично реагирует на клоназепам, хотя реакция не столь выражена, как на алкоголь (который может купировать симптомы). В одном сообщении [Priori et al. 2000] чувствительную к алкоголю миоклоническую дистонию успешно лечили 6,125 гр гамма-гидроксибутировой кислоты ежедневно. В тяжелых случаях миоклоус-дистонии может помочь двусторонняя глубокая стимуляция головного мозга.
Спинальный миоклонус. Фармакологическое лечение спинальной формы оставляет желать лучшего. Клоназепам является препаратом выбора для обоих типов спинального миоклонуса, а доза < 6 мг необходима для уменьшения выраженность сегментарного миоклонуса. Леветирацетам также может оказаться полезным в его терапии.
Сегментарный миоклонус. Сегментарный миоклонус, независимо от его происхождения (тремор неба, сегментарный спинальный миоклонус), поддается ботулинотерапии, но с переменным успехом.
Периферический миоклонус. Фармакологическое лечение малоэффективно, однако, в некоторых случаях может помочь прием карбамазепина и инъекции ботулинического токсина.
Тики.
Тики — это повторные, стереотипные, насильственные движения. Тики, длящиеся более одного года с одними лишь моторным компонентом, обозначаются как хронические моторные тики, а те, у которых только вокальные проявления — как хронические вокальные тики. Это различие не имеет строгого нейробиологического обоснования, учитывая, что и те, и другие являются продуктом двигательной активности на уровне мышц (в случае вокальных тиков — мышц гортани, глотки или дыхательных мышц). Транзиторными же являются те, которые длятся менее одного года.
Синдром Жиля де ля Туретта (СЖТ) — патология развития нервной системы, затрагивающая 1 % населения, которая характеризуется множественными моторными и вокальными тиками с дебютом в детском возрасте [36].
Синдром Туретта является наиболее часто встречаемым наследственным по неменделевскому типу нейропсихиатрическим заболеванием (соответствующий семейный анамнез — у 52 % пациентов; 10-кратное увеличение вероятности развития заболевания — среди родственников первого поколения, по сравнению с общепопуляционной; отношение конкордантности среди монозиготных близнецов — 5:1, по сравнению с дизиготными). Однако еще не удалось обнаружить определенных генов, нарушение в деятельности которых ответственно за развитие заболевания. СЖТ представляет собой мультифакториальное заболевание, развитие которого обуславливает сочетание генетических изменений и влияния окружающей среды во время пре- и постнатального развития.
Моторные и вокальные тики включают три компонента:
1. Усиливающееся чувство необходимости реализовать движение, вплоть до ощущения физической боли, зуда и дискомфорта;
2. Непосредственное осуществление тика;
3. Ощущение облегчения после его осуществления.
Пациенты с СЖТ в состоянии контролировать тики, предотвращая их реализацию, однако чем дольше человек сдерживается, тем сложнее ему это дается. Скопившийся дискомфорт за день, например, при подавлении тиков в школе, может (и должен) реализоваться дома. Это отличает СЖТ от других гиперкинезов.
Механизмы, вовлеченные в развитие данного заболевания лежат в межнейрональных взаимосвязях между базальными ганглиями, таламусом и корой больших полушарий.
Антипсихотики могут снижать выраженность симптоматики, однако к лекарственным средствам прибегают нечасто, предпочитая когнитивно-поведенческую терапию.
Тяжесть в состояние пациента также вносят сопутствующие психиатрические заболевания: синдром дефицита внимания и гиперактивности (СДВГ), обсессивно-компульсивное расстройство (ОКР), депрессия, расстройства аутистического спектра (РАС), генерализованное тревожное расстройство, оппозиционно-вызывающее расстройство (ОВР).
Критерии диагностики синдрома Туретта. Моторные тики — внезапные, быстрые, повторяющиеся, неритмичные, стереотипные движения. Тики могут проявиться в любой части тела, но чаще встречаются в области головы и шеи. Вокальные тики выражаются формированием любого звука: шмыгание носом, кряхтение, жужжание, щелканье, повторение слов. Копролалия встречается только в 10 % случаев пациентов с СЖТ.
Критерии согласно DSM-5:
• Множественные моторные и один или более вокальных тиков присутствуют в течение некоторого времени заболевания, не обязательно одновременно;
• Частота проявления тиков может возрастать и снижаться, но они сохраняются в течение более одного года с момента появления первого тика;
• Начало до 18 лет;
• Нарушение не связано с другими заболеваниями или приемом препаратов.
Заболевание обычно дебютирует к 4–6 годам, постепенно нарастая к 10–12. Начало острое, тики повторяются множество раз в течение дня. Симптоматика усиливается в течение нескольких дней и недель, после чего выходит на плато (недели — годы), дабы затем начать регрессировать. Длительность прогрессии и плато вариабельна для каждого пациента.
Дифференциальная диагностика тиков. Абсанс-припадки с миоклонусом век — начинаются в том же возрасте, что и СЖТ, при этом тики могут манифестировать с век, что делает эти заболевания схожими. Однако моторные тики не сопровождаются нарушением сознания и меньше длятся. Для абсанс-эпилепсии, кроме того, характеры особые изменения на ЭЭГ, которые не наблюдается при тиках.
Стереотипии — редкие, ритмичные, повторные, фиксированные, предсказуемые движения, наблюдающиеся среди детей с нормальным развитием. Примерами являются взмахи руками, кистями, кивание головой, покачивания вперед и назад. В отличии от СЖТ, они обычно начинаются до 3-х лет, часто во младенческом возрасте. Стереотипии проявляются однократными повторяющимися движениями, в отличии от множества моторных тиков. Их проще контролировать, и возникают они, как правило, при волнении. Полностью регрессируют к младшему школьному возрасту.
Хорея характеризуется резкими непроизвольными движениями, в частности в области плечевого пояса, бедер и лица. Движения беспорядочны, но постоянны, в отличие от моторных тиков. Хорея гораздо реже встречается в детском возрасте.
Пароксизмальная дискинезия — гиперкинезы часто провоцируются стартл-рефлексом или резким движением. Приступы, как правило, короткие, длятся всего несколько секунд или минут. Симптомам могут предшествовать необычные ощущения в конечностях. Часто сопутствует дистония, синдромы хорея-дистония, баллизм-дистония. Наследование аутосомно-доминантное, но может быть спорадическим.
Акатизия характеризуется ощущением внутреннего беспокойства и неспособности оставаться на месте. Обычно она является побочным эффектом антипсихотиков, антидепрессантов или противорвотных препаратов. Как правило, акатизия встречается у взрослых, нередко и у детей.
Обсессивно-компульсивное расстройство (ОКР). Кроме того, что дифференциальная диагностика тиков и ОКР довольно-таки затруднительна, сами эти заболевания часто сопутствуют друг другу. Движения при ОКР необходимо осуществить из-за беспокойства и навязчивых мыслей («Если я не сделаю это вот так, произойдет что-то ужасное»). Моторным тикам предшествуют скорее бессознательное беспокойство и внутренний дискомфорт.
Заболевания, ассоциированные с развитием вторичных тиков:
1. Нарушения развития нервной системы:
— Умственная отсталость;
— Расстройства аутистического спектра (включая синдром Аспергера);
— Синдром Ретта;
— Заикание (невротическая форма);
— Генетические и хромосомные аномалии:
• Х-сцепленная умственная отсталость (MRX23);
• Наследственная остеодистрофия Олбрайта;
• Мышечная дистрофия Дюшенна;
• Гемофилия А (VIII фактор);
• Синдром ломкой X-хромосомы;
• Синдром Леша-Нихана;
•Трисомия X и 9p мозаицизм;
• 47 XXY кариотип;
• Частичная трисомия 16;
• 9p моносомия;
• Синдром Беквита-Видемана;
• Туберозный склероз;
• Врожденная гиперплазия надпочечников вследствие дефицита 21-гидроксилазы;
• Фенилкетонурия;
• Дисгенезия мозолистого тела;
• Краниосиностозы;
• Синдром Клайнфельтера;
• Нейрофиброматоз;
2. Острые поражения головного мозга:
— Посттравматические;
— Сосудистые;
— Инфекционные:
• Вирус герпес зостер;
• Вирус простого герпеса;
• Mycoplasma pneumoniae;
• Болезнь Лайма;
— Постинфекционные:
• Педиатрическое аутоиммунное нервно-психическое расстройство, связанное со стрептококковой инфекцией (PANDAS)
3. Нейродегенеративные заболевания:
• Болезнь Хантингтона;
• Синдромы нейроакантоцитоза;
• Нейродегенерация с накоплением железа в мозге;
4. Другие системные заболевания:
• Синдром Бехчета;
• Антифосфолипидный синдром;
5. Травма периферической нервной системы;
6. Лекарства и токсины:
• Амфетамин;
• Кокаин;
• Героин;
• Метилфенидат;
• Премолин;
• Антипсихотики (блокаторы D2-рецепторов) — редко могут приводить к тардивным тикам:
- Флуфеназин;
- Перфеназин;
- Тиотиксен;
• Антидепрессанты;
• Противоэпилептические препараты:
- Карбамазепин;
- Фенитоин;
- Фенобарбитал;
- Ламотрижин;
• Леводопа.
7. Функциональные тикоподобные рывки [39].
Терапевтическое вмешательство при первичных тиках зачастую ограничивается психотерапией. Из фармакологических препаратов применяются антипсихотики и терапия сопутствующих психиатрических заболеваний. В рефрактерных к терапии случаях можно рассматривать возможность применения инъекций ботулотоксина.
Тремор
Тремор — это ритмичные, колебательные, вынужденные движения частей тела, продуцированные переменным сокращением реципрокно иннервируемых мышц [38].
Описание тремора основывается на следующих параметрах:
• Возникает в покое (тремор покоя) или при движении (кинетический тремор);
• Локализация тремора;
• Частота колебаний;
• Ритмичность;
• Амплитуда колебаний;
• Индуцирующие или нивелирующие факторы (примеры препаратов и токсинов, способных индуцировать тремор в табл. 8);
• Другая неврологическая симптоматика.
Основные виды тремора. Тремор покоя — спонтанная мышечная активность, наблюдаемая в покое, когда пациент не совершает какого-либо действия либо не поддерживает вовлеченную часть тела против гравитации (например, кладет руки на стол, колени). Может усиливаться при отвлечении на другие задачи, например, во время беседы.
Двигательный тремор (акционный) — любой тремор, продуцированный сокращением мышц:
•Постуральный тремор — возникающий в результате сопротивления гравитации. Реже тремор может индуцироваться определенной позой;
• Кинетический тремор — любой тремор, вызванный сокращением мышц;
• Неспецифический кинетический тремор — тремор, формирующийся при любом движении без определенной цели;
• Интенционный тремор — активируется при попытке осуществить определенное действие (например, коснуться пальцем до кончика носа). Чем ближе цель, тем выше амплитуда тремора;
•Специфический кинетический тремор — появляется только в случае определенного действия.
Особые виды тремора. Также выделяют форму дистонического тремора — это аритмичные (интервалы между следующими друг за другом движения отличаются) и/или нерегулярные (различия в амплитудах) колебания, которые, по сути, не являются истинным тремором. Как уже было описано, тремор этот обусловлен сокращением мышц агонистов и антагонистов пораженной конечности, в результате чего и формируются эти колебательные движения.
Нейропатический тремор (НТ) часто определяют как «эссенциально-подобный тремор». НТ и эссенциальный тремор (ЭТ) оба, как правило, постуральные и кинетические, преимущественно в дистальных отделах конечностей, могут иметь одинаковую амплитуду и частоту. НТ чаще проявляется как нерегулярный, нежели ЭТ. Как НТ, так и атипичные и тяжелые случаи ЭТ могут демонстрировать тремор покоя, который имеет меньшую амплитуду, нежели постуральный и кинетический.
Тремор Холмса (среднемозговой, рубральный тремор) характеризуется наличием комбинированного тремора (покоя и интенционного), чаще всего локализующегося в верхних конечностях, который ассоциирован с ипсилатеральной дисметрией и дисдиадохокинезом. Вдобавок, может наблюдаться постуральный тремор в проксимальных отделах конечностей. Его частота < 4,5 Гц и может быть нерегулярной. Причина тремора Холмса — повреждение одновременно дофаминергической системы и связей мозжечка с таламусом или нижними ядрами оливы. Тремор формируется спустя некоторое время после поражения (от 1 до 24 месяцев). Этиологией чаще всего выступает стволовой инсульт или травма.
Физиологический тремор — тремор, возникающий у здоровых людей (точнее сказать, при отсутствии заболеваний, способных индуцировать тремор), на фоне тревоги, стресса, приема стимуляторов.
Другой вид тремора, эссенциальный, обсуждается ниже.
Частота колебаний. В большинстве клинических случаев тремора частота колебаний составляет от 4 до 12 Гц. Тремор при болезни Паркинсона имеет частоту 3–5 Гц, эссенциальный тремор — 5–10 Гц. Кроме болезни Паркинсона, тремор с частотой < 4 Гц проявляется при мозжечковых заболеваниях, треморе Холмса, треморе мягкого неба и в случае побочных реакций и интоксикации некоторыми препаратами. Первичный ортостатический тремор характеризуется частотой 12–18 Гц.
Частоту колебаний правильно вычислять с помощью ЭНМГ. Однако расхождение клинической картины тремора с общепринятой частотой не является исключающим определенное заболевание критерием.
Таблица 8. Препараты и токсины, вызывающие тремор.
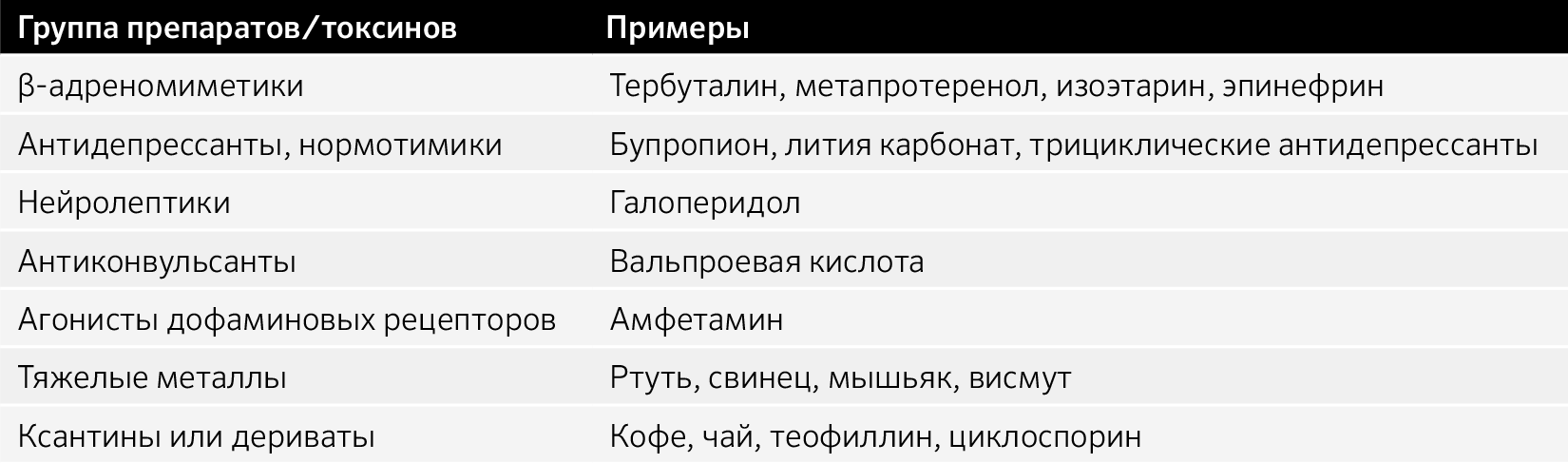
Побочные эффекты препаратов в виде тремора имеют дозозависимый эффект. У пациентов, получающих литий или вальпроат натрия, при развитии тремора должны быть исследованы уровни концентрации этих веществ в крови. Употребление кофе, чая или других стимуляторов в необычно высоких количествах может являться достаточным для появления тремора. Гипертиреоз и гипогликемия также могут индуцировать тремор.
Эссенциальный тремор
ЭТ на данный момент принято считать нейродегенеративным заболеванием. Данный вид тремора является наиболее распространенным (не считая физиологического). Это кинетический, постуральный, интенционный, реже — тремор покоя, вовлекающий верхние конечности, шею, голосовой аппарат или челюсть. Кроме того, с ЭТ часто ассоциируются другие неврологический и нейропсихиатрические состояния (т. н., «ЭТ-плюс»: атаксия, дистония, когнитивные изменения, паркинсонизм). ГАМК-ергические препараты и β-блокаторы имеют ограниченный эффект на прогрессирование заболевания. Вносят вклад в развитие ЭТ как окружающие факторы (свинец, гармалин и т. д.), так и генетическая детерминированность (вероятность развития болезни среди родственников первого поколения и монозиготных выше в сравнении с гетерозиготными близнецами) [40].
Основные патофизиологические аспекты заболевания вращаются вокруг треугольника Молляре (triangle of Guillain and Mollaret) (рис. 8), ядер нижней оливы и клеток Пуркинье. Главной функцией треугольника Молляре является настройка точных, аккуратных движений. Ядра нижней оливы (ЯНО), входящие в состав этого анатомического образования, формируют внутренний ритм движения.
Исследования увеличения активности нижней оливы (при использовании гармалина) продемонстрировали повышение активации клеток Пуркинье через лиановидные волокна. Стимуляция в итоге приводит к синхронизации ритма, что приводит к тремору. Предполагалось, что нейроны ЯНО относятся к пейсмейкерам, однако накапливающиеся исследования пациентов с ЭТ демонстрировали сохранность и непричастность ЯНО к развитию патологии, в отличие от дегенерации мозжечка. Конечно, это не исключает нижнюю оливу из патофизиологических аспектов тремора, однако ясно, что ЯНО не являются ключевым аспектом [40]. Большее внимание сосредотачивается на клетках Пуркинье, а ЭТ предлагают переименовать в «дегенеративную Пуркиньепатию» [43].
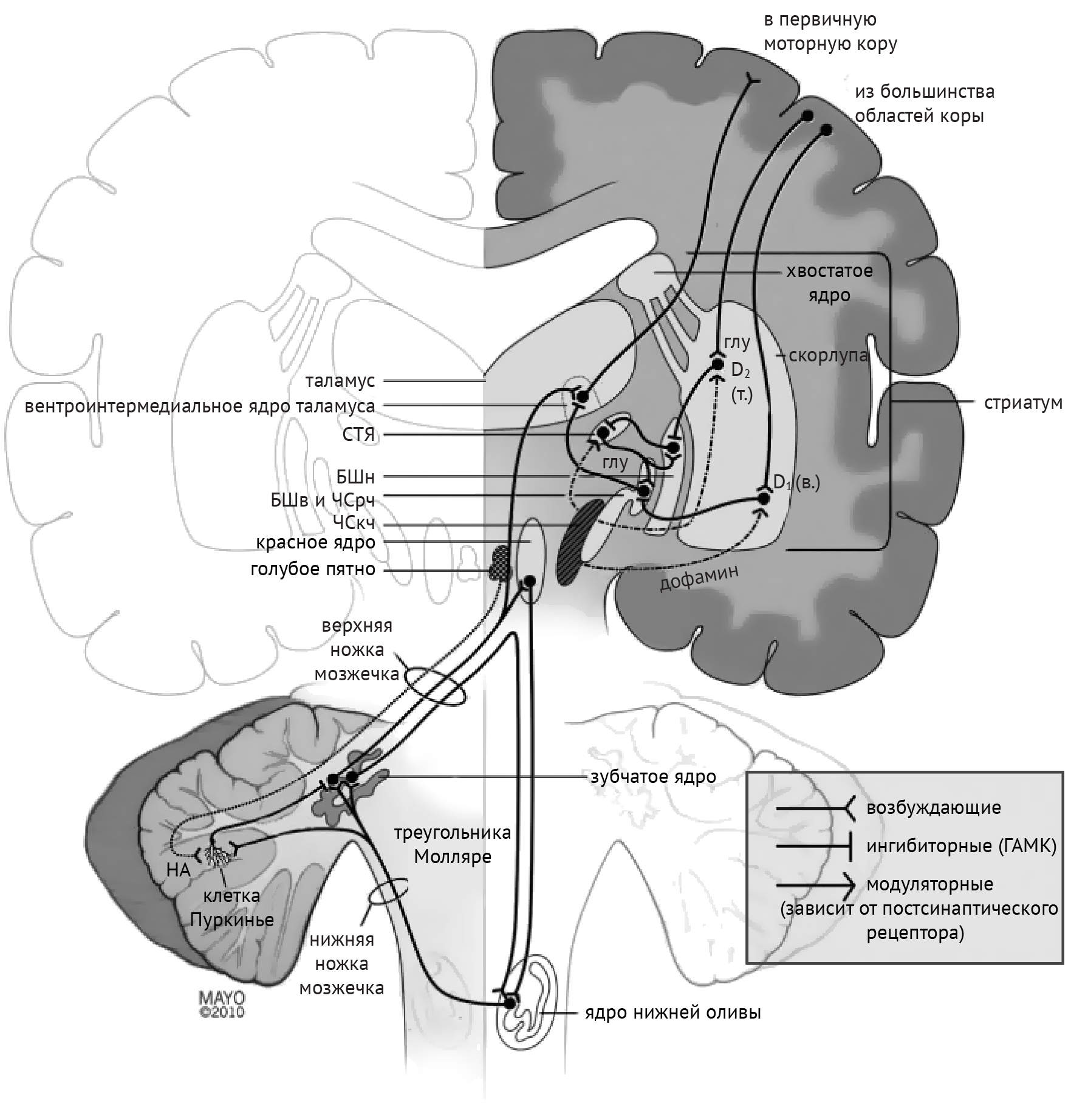
Дегенеративный каскад ЭТ патоморфологически развивается по следующему типу (табл. 9).
Таблица 9. Патоморфологические стадии прогрессирования дегенерации мозжечка при ЭТ.
Кроме поражения нейронов Пуркинье, корзинчатых нейронов, зубчатого ядра и их аксонов и дендритов, в патологию вовлекается и макроглия. Так, у пациентов с ЭТ наблюдается снижение экспрессии EAAT2 (транспортер возбуждающих аминокислот 2 типа, или транспортер глутамата 2 типа), приводящее к увеличению количества глутамата в межсинаптической щели и повышению вероятности эксайтотоксичности клеток Пуркинье.
Синаптический рецидивирующий спрутинг ГАМК-ергических вставочных нейронов ясен: снижение уровня ГАМК может привести к дегенерации клеток Пуркинье, что, в свою очередь, приведет к необходимости усилить ГАМК-ергическое влияние через разрастание сетей проекций от корзинчатых интернейронов, коллатералей от клеток Пуркинье с целью предотвращения эксайтотоксичности. Это приводит к еще меньшей экспрессии ГАМК нейронами Пуркинье, что путем обратной связи увеличивает межсинаптические контакты лиановидных волокон с дендритами клеток Пуркинье, выдавая порцию глутамата. Порочный цикл повторяется вновь. Изменения, вносящие существенное снижение ГАМК-ергического влияния, называются «идеальный шторм» (рис. 9).
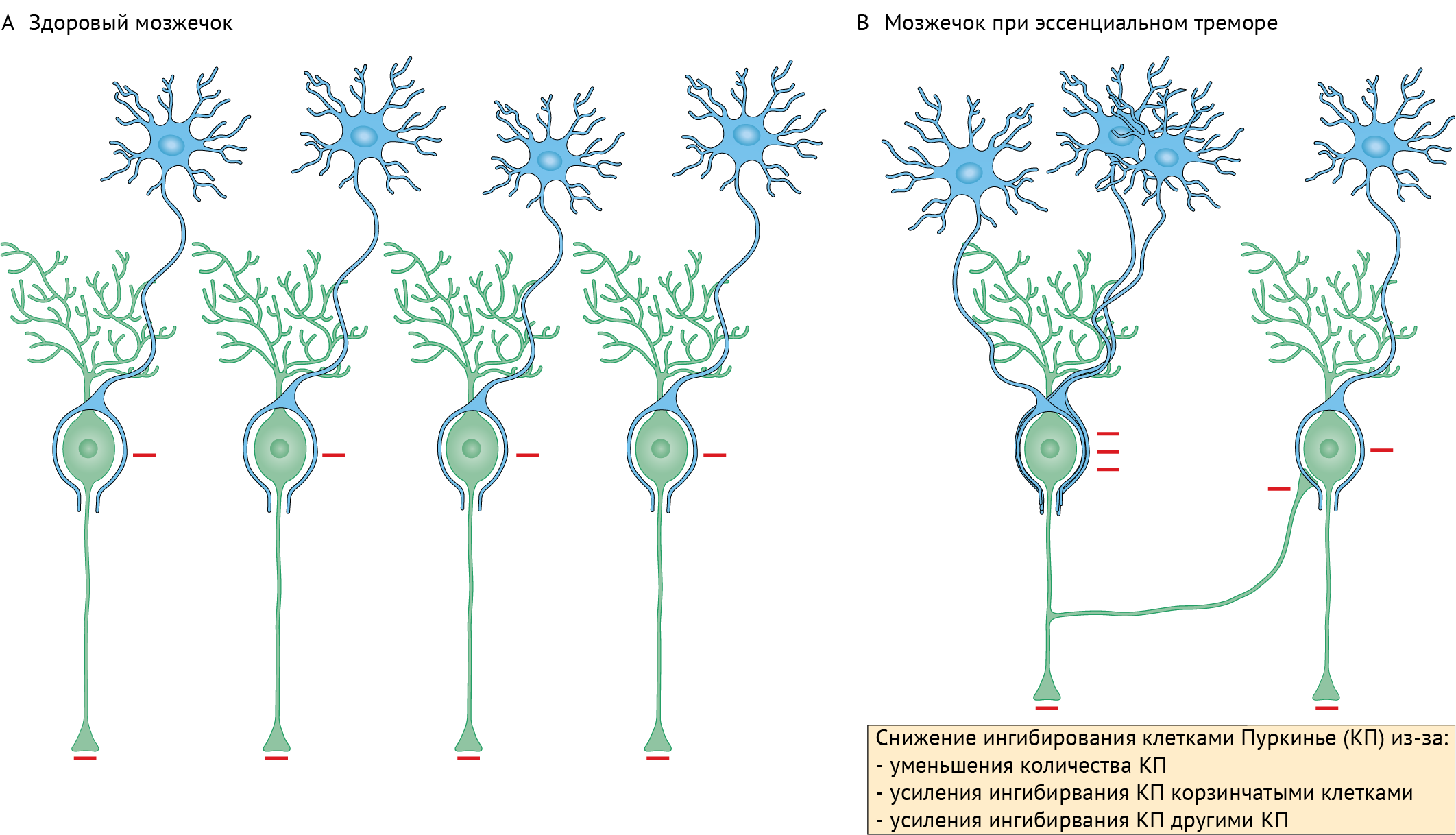
б | В мозжечке у людей с эссенциальным тремором наблюдаются потеря клеток Пуркинье, усиление коллатерального спрутинга и гипертрофия аксональных ответвлений корзинчатых нейронов. Конечным результатом этих трех изменений является снижение ингибирующего ГАМК-ергического влияния коры мозжечка на его ядра. Наблюдаемое состояние гипервозбуждения может являться причиной развития тремора.
Именно патологическая синхронизация в системе связей между клетками Пуркинье — ядрами мозжечка — ЯНО вносит существенный вклад в реализацию тремора. Как уже было сказано, нейроны ядер оливы не являются пейсмейкерами, однако наличие между ними плотных контактов способствуют проведению синхронизированных разрядов.
Немного о насущном
К вопросу о глубокой стимуляции мозга и гипо-, гиперкинезам. Глубокая стимуляция головного мозга (ГСМ) — метод, основанный на инвазии электродов в определенные структуры мозга для мягкого воздействия электрического тока на них. Данный метод используется для лечения заболеваний, резистентных к консервативной терапии: психиатрических (РАС, депрессия) и неврологических заболеваний, с целью смягчение двигательных и иных нарушений (болезнь Паркинсона, генерализованная и сегментарная формы дистонии, ЭТ, эпилепсия и др). Данный метод представляет собой нейрохирургическую операцию, поэтому сохраняет за собой опасности оперативного вмешательства. Также стоит учитывать необходимость осуществления операции по замене источника питания электродов раз в 3–4 года. ГСМ несет в себе огромные возможности по лечению психиатрических и неврологических заболеваний и изучению нейрофизиологии головного мозга. Тем не менее, лечение, даже при отсутствии осложнений, может иметь множество побочных явлений [44, 45].
Терапия болезни Паркинсона основана на десинхронизации патологических паттернов, сформированных в результате нарушения нормального проведения сигнала. Поражение дофаминергических нейронов (например, дегенерация > 80 % черной субстанции), проявляется возникновением патологического β-ритма в базальных ганглиях (15–30 Гц). В результате воздействия электрода на субталамическое ядро, происходит билатеральная стимуляция последнего, что приводит к подавлению высоких уровней β-ритма колебаний головного мозга (конечно, не исключено влияние стимуляции на соседние структуры ГМ). Данный метод позволяет буквально программировать передачу синаптических сигналов. Тем не менее, микроскопическая хирургическая травматизация субталамического ядра при инвазии датчика зачастую вызывает нарушения когнитивных функций, в частности — уменьшение словарного запаса, ухудшение спонтанности речи, другие специфические психические отклонения. Недавно проведенные исследования демонстрируют также наличие психофизиологического дефицита у пациентов, длительное время подвергавшихся высокочастотной стимуляции (> 130 Гц), приводящей к нарушению взаимодействия базальных ганглий и фронтальных долей головного мозга. В отличие от высокочастотной, низкочастотная стимуляция головного мозга (< 130 Гц) достоверно приводит к увеличению вербальных функций пациента, активируя фронтальные пути. Индивидуальное программирование под каждого пациента способно минимизировать побочные явления [44].
Следующим неприятным фактом будет являться необходимость операционного вмешательства для замены батареи, которой при постоянной стимуляции хватает лишь на 3–4 года терапии. Непостоянная стимуляция приводит к возвращению симптомов болезни. Стимуляция же в режиме нон-стоп приводит к ухудшению когнитивных функций, нарушению речи, возобновлению непроизвольных движений. К счастью, существуют способы, позволяющие титровать нагрузку на нейроны. В данный момент уже созданы несколько вариантов адаптивной ГСМ, в которых режим стимуляции меняется в ответ на физиологические сигналы.
О самих вариантах вы можете прочитать на http://neuronovosti.ru/that-strange-dbs/.
Литература:
- Dale Purves Neuroscience №6 2018 (416-425);
- Balint, B., Mencacci, N.E., Valente, E.M. et al. Dystonia. Nat Rev Dis Primers 4, 25 (2018). doi.org/10.1038/s41572-018-0023-6
- Tanabe, L., Kim, C., Alagem, N. et al. Primary dystonia: molecules and mechanisms. Nat Rev Neurol 5, 598–609 (2009). doi.org/10.1038/nrneurol.2009.160
- Bhidayasiri R., Tarsy D. (2012) Cervical Dystonia: Sensory Tricks. In: Movement Disorders: A Video Atlas. Current Clinical Neurology. Humana, Totowa, NJ. doi.org/10.1007/978-1-60327-426-5_43
- Cox BC, Cincotta M, Espay AJ. Mirror movements in movement disorders: a review. Tremor Other Hyperkinet Mov (N Y). 2012;2:tre-02-59-398-1. doi:10.7916/D8VQ31DZ
- Albanese A, Sorbo FD. Dystonia and Tremor: The Clinical Syndromes with Isolated Tremor. Tremor Other Hyperkinet Mov (N Y). 2016;6:319. Published 2016 Apr 5. doi:10.7916/D8X34XBM
- Horisawa S, Taira T, Goto S, Ochiai T, Nakajima T. Long-term improvement of musician's dystonia after stereotactic ventro-oral thalamotomy. Ann Neurol. 2013;74(5):648-654. doi:10.1002/ana.23877
- Termsarasab P. Chorea [published correction appears in Continuum (Minneap Minn). 2019 Dec;25(6):1834]. Continuum (Minneap Minn). 2019;25(4):1001-1035. doi:10.1212/CON.0000000000000763
- Ivan Donaldson, C. David Marsden, Susanne Schneider, and Kailash Bhatia. Marsden's Book of Movement Disorders. Oxford University Press, 2012.
- John G.L Morris DM, et al. Athetosis II: The syndrome of mild athetoid cerebral palsy. Movement Disorders. Vol. 17, No. 6, 2002, pp. 1281–1287.
- Opal P, Ashizawa T. Spinocerebellar Ataxia Type 1. 1998 Oct 1 [Updated 2017 Jun 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Pulst SM. Spinocerebellar Ataxia Type 2. 1998 Oct 23 [Updated 2019 Feb 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Paulson H, Shakkottai V. Spinocerebellar Ataxia Type 3. 1998 Oct 10 [Updated 2020 Jun 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Carroll LS, Massey TH, Wardle M, Peall KJ. Dentatorubral-pallidoluysian Atrophy: An Update. Tremor Other Hyperkinet Mov (N Y). 2018;8:577. Published 2018 Oct 1. doi:10.7916/D81N9HST
- Batla A, Adams ME, Erro R, et al. Cortical pencil lining in neuroferritinopathy: a diagnostic clue. Neurology. 2015;84(17):1816-1818. doi:10.1212/WNL.0000000000001511
- Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. 2002 Jul 30 [Updated 2018 Mar 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Velayos Baeza A, Dobson-Stone C, Rampoldi L, et al. Chorea-Acanthocytosis. 2002 Jun 14 [Updated 2019 Apr 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Członkowska, A., Litwin, T., Dusek, P. et al. Wilson disease. Nat Rev Dis Primers 4, 21 (2018).doi.org/10.1038/s41572-018-0018-3
- Schneider, S., Walker, R. & Bhatia, K. The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test. Nat Rev Neurol 3, 517–525 (2007). doi.org/10.1038/ncpneuro0606
- https://emedicine.medscape.com/article/1150420-overview
- https://emedicine.medscape.com/article/1113394-overview#a4
- Coutinho P, Barbot C. Ataxia with Oculomotor Apraxia Type 1. 2002 Jun 11 [Updated 2015 Mar 19]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Moreira MC, Koenig M. Ataxia with Oculomotor Apraxia Type 2. 2004 Nov 15 [Updated 2018 Jul 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49-71. doi:10.1016/B978-0-12-410502-7.00003-X
- Miyajima H, Hosoi Y. Aceruloplasminemia. 2003 Aug 12 [Updated 2018 Sep 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Larson A, Goodman S. Glutaric Acidemia Type 1. 2019 Sep 19. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Shchelochkov OA, Carrillo N, Venditti C. Propionic Acidemia. 2012 May 17 [Updated 2016 Oct 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Jung HH, Danek A, Walker RH, et al. McLeod Neuroacanthocytosis Syndrome. 2004 Dec 3 [Updated 2019 May 23]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Jinnah HA. HPRT1 Disorders. 2000 Sep 25 [Updated 2020 Aug 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Evidente VGH. X-Linked Dystonia-Parkinsonism. 2005 Dec 13 [Updated 2018 Feb 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. 2003 Oct 30 [Updated 2017 Sep 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [Updated 2018 Nov 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
- Hyperkinetic Movement Disorders by Roger M Kurlan, Paul E Greene, Kevin M Biglan. Oxford University Press 2015. ISBN 978–0–19–992564–3
- Zutt, R., van Egmond, M., Elting, J. et al. A novel diagnostic approach to patients with myoclonus. Nat Rev Neurol 11, 687–697 (2015). doi.org/10.1038/nrneurol.2015.198
- Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4(1):47-62. doi:10.1177/1756285610395653
- Martins WA, Schilling LP, Neto FK, Becker J. Hypertrophic Olivary Degeneration Secondary to Neuro-Behçet's Disease. Clin Neuroradiol. 2016;26(1):99-102. doi:10.1007/s00062-015-0384-0
- Eberhardt O, Topka H. Myoclonic Disorders. Brain Sci. 2017;7(8):103. Published 2017 Aug 14. doi:10.3390/brainsci7080103
- Jones KS, Ramphul K. Tourette Syndrome And Other Tic Disorders. [Updated 2020 Jun 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan.
- Martino D, Mink JW. Tic disorders. Continuum (Minneap Minn). 2013;19(5 Movement Disorders):1287-1311. doi:10.1212/01.CON.0000436157.31662.af
- Puschmann A, Wszolek ZK. Diagnosis and treatment of common forms of tremor. Semin Neurol. 2011;31(1):65-77. doi:10.1055/s-0031-1271312
- Louis, E.D., Faust, P.L. Essential tremor pathology: neurodegeneration and reorganization of neuronal connections. Nat Rev Neurol 16, 69–83 (2020). doi.org/10.1038/s41582-019-0302-1
- Louis, E.D., Diaz, D.T., Kuo, S. et al. Inferior Olivary nucleus degeneration does not lessen tremor in essential tremor. cerebellum ataxias 5, 1 (2018). doi.org/10.1186/s40673-018-0080-3
- Grimaldi G, Manto M. Is essential tremor a Purkinjopathy? The role of the cerebellar cortex in its pathogenesis. Mov Disord. 2013;28(13):1759-1761. doi:10.1002/mds.25645
- Pham T, Bronstein JM. Neuropsychological outcomes from deep brain stimulation-stimulation versus micro-lesion. Ann Transl Med. 2017;5(10):217. doi:10.21037/atm.2017.02.16
- Bartrés-Faz D, Rossi S, Pascual-Leone A. Editorial: Non-invasive Brain Stimulation and Plasticity Changes in Aging. Front Aging Neurosci. 2016;8:96. Published 2016 May 4. doi:10.3389/fnagi.2016.00096
- «Электростимуляция глубоких структур головного мозга при экстрапирамидных заболеваниях. Принципы программирования» А.А. Гамалея, А.А. Томский, Е.В. Бриль, В.А. Шабалов. «Нервные болезни» 4*2012
