
Патологические включения при нейродегенеративных заболеваниях (часть 1)
Небольшой дисклеймер: ниже речь пойдет об общих и частных механизмах образования патогенных белков при различных нейродегенеративных заболеваниях (НДЗ), а не о самих НДЗ (этиологии, диагностике, симптомах и далее по списку). Оптимально употреблять в сочетании с другими источниками о конкретных заболеваниях для полноты картины.
Для начала разберемся с терминологией. Слово «нейродегенеративное» подразумевает, что данное заболевание сопровождается медленно прогрессирующим необратимым снижением числа нейронов. Акцент на «медленно прогрессирующим», поскольку загубить нейроны — дело нехитрое, и справляется с этим целый спектр состояний — от инсульта и ЧМТ до идущих под руку в ближайшую рюмочную алкоголизма и печеночной энцефалопатии. Условно к нейродегенеративным относят две группы заболеваний — деменции и двигательно-дегенеративные расстройства. Условно, потому что зачастую НДЗ совмещают приятное с полезным и даже плавно перетекают одно в другое (см. ALS/FTLD).
Уже на ранних этапах изучения НДЗ были выявлены три характерные для них черты: прогрессирующая гибель нейронов, астроглиоз (рубцевание участков погибших нейронов за счет астроцитов) и отложение патологических белков с измененными физико-химическими характеристиками. Такие белки, в свою очередь, подразделяются на вне- и внутриклеточные; последние могут скапливаться в различных частях клеток нервной системы, в т. ч. и внутри ядра (Рис. 1). И, да, клеток нервной системы вообще, а не только нейронов. Как в том анекдоте: было больно, зато всем досталось.
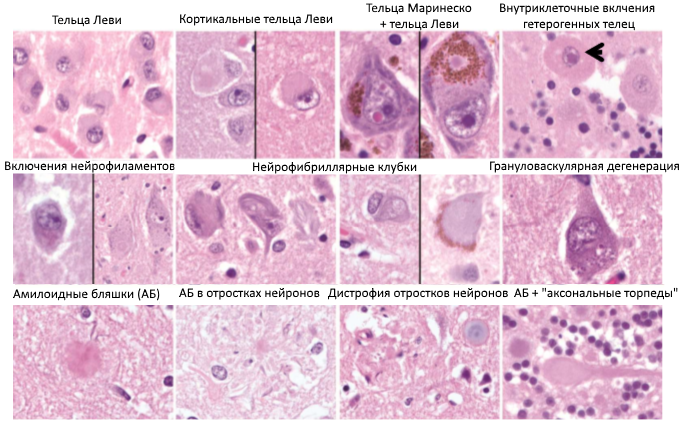
Рисунок 1 | Возможные виды патологических включений
Прежде чем приступить к разбору механизмов образования подобных включений, вспомним курс биохимии, а именно уровни структурной организации белков. Нас, в частности, интересуют регуляторные вторичные структуры. Принципиально на этом уровне белок либо закручивается в спираль (почти всегда — α-спираль), либо ложится слоями — такая структура называется β-листами (Рис. 2). Остальные возможные конфигурации редки и представляют собой вариации на тему спирали.
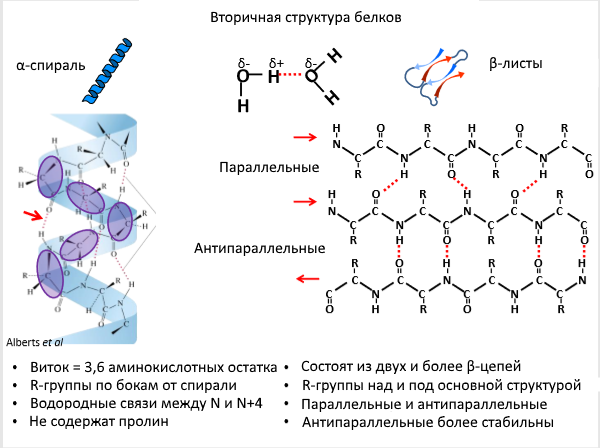
Рисунок 2 | Вторичная структура белков
Попробуем описать общий механизм образования подобных включений с высоты этих знаний. Нормальный белок со вторичной α-спиралевидной структурой, существующий в нервной системе, под действием генетических и внешних факторов в определенный момент укладывается неправильно, образуя β-цепи. Те, в свою очередь, накапливаются, образуя все большие конгломераты β-листов (и, возможно, способны вовлекать в этот процесс неизмененные α-спирали, вынуждая принять форму β-цепей; на этом предположении основана одна из ведущих прионных теорий, но о ней в другой раз) (Рис. 3). С увеличением количества подобных скоплений приумножаются и проблемы.
Важное уточнение: измененные белки продолжают структурную организацию, имеют третичную и четвертичную структуры, однако нарушение кроется именно на этапе вторичной.

Рисунок 3 | Процесс формирования патологических включений при НДЗ
Итак, о проблемах. С изменением конфигурации утрачивается и функция белка. Так, патологический тау-белок не способен стабилизировать микротрубочки, что нарушает аксональный транспорт. Аксон, как известно, заканчивается синапсом, и если последний не получает ресурсов для осуществления деятельности, он эту деятельность сворачивает. Эффект домино набирает обороты: следующий в дуге нейрон, перестав получать сигналы, подвергается атрофии и гибнет. И, наконец, круг замыкается: с гибелью постсинаптических звеньев цепи наш изначально пораженный нейрон также становится бесполезен и атрофируется. Доживет ли он до этого момента — другой вопрос.
Дело в том, что измененный белок не только утрачивает полезную функцию, но и может приобрести новую, вредоносную. Продолжаем пример с тау-белком: патологический вариант бесполезен и нерастворим, и при этом создает все большие скопления на поверхности микротрубочек. Участки с таким «покрытием» что российские трассы: едешь себе спокойно по ямам, а потом — бац! — дорога. Спойлер: до «бац!» доезжают не все. Таким образом, на обочине микротрубочек начинает накапливаться не доставленный к синапсам груз из химически активных молекул и остатков митохондрий. Подобный «мусор» ставит нейрону вилку: либо тратишь ресурсы на его переработку, либо миришься с нарастающей токсичностью.
Даже если прямого вреда от конгломератов патологических белков нет, они все равно занимают пространство. Если речь о внутриклеточных отложениях, рано или поздно они до такой степени затрудняют функционирование, что клетка гибнет (нет, до «она лопнула» не доходит). «Насильственная» гибель клетки (не за счет апоптоза) сопровождается высвобождением химически активных молекул и чревата воспалительным процессом. Высвобождаются, кстати, и скопления белков, становясь таким образом внеклеточными. Это не отменяет возможности отложения сразу вне клетки. Казалось бы, в этом случае белок практически безвреден: он не подрывает деятельность клетки изнутри, и вряд ли достигнет размера, необходимого для нарушения функций ЦНС, как это могло бы сделать объемное образование.
Это не так. Внеклеточные конгломераты, во-первых, меняют онкотическое давление среды, «стягивая» на себя жидкость и тем самым влияя на обменные процессы в окружающих клетках. Во-вторых, как и в любых белках, в них есть группы с различными зарядами, что изменяет электрофизические свойства экстрацеллюлярного пространства, нарушая электролитный баланс, ионный обмен и потенциал действия нервных клеток. В-третьих, подобные изменения могут повысить проницаемость гемато-энцефалического барьера, что чревато проникновением в нейроны целого спектра нежелательных веществ, клеток (лейкоциты будут не против попытаться разрушить патологический белок) и даже патогенных микроорганизмов. В-последних, «внеклеточным» является и ликвор, и в нем подобные изменения еще критичнее.
А теперь перейдем к конкретным белкам. Для более простого ориентирования на местности пользуйтесь схемой ниже; она же позволит оценить все многообразие вызываемых НДЗ.
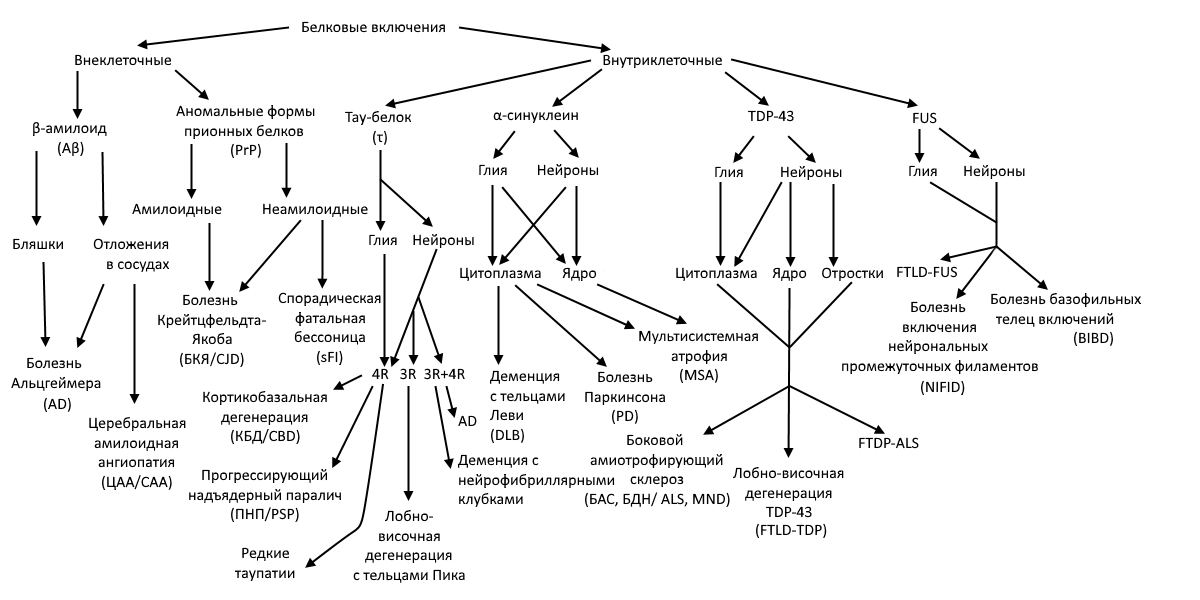
Рисунок 4 | Классификация протеинопатий ЦНС. В скобках — общепринятые сокращения в русскоязычной/англоязычной литературе, чтобы облегчить поиск литературы для всех заинтересовавшихся :)
β-амилоид (Аβ)
Под этим термином расположилась группа пептидов, образовавшихся из белка-предшественника в результате воздействия β- и γ-секретазы. Патогенность определяется количеством аминокислот в составе пептида: так, варианты с 39/40 остатками менее патогенны и накапливаются в сосудах (ряд исследователей считает Аβ40 вообще не патогенным), в то время как 42- и 43-аминокислотные формы более агрессивны и образуют бляшки. Следует отметить, что амилоидоз в целом — состояние, не уникальное для ЦНС, и имеющее определенный генетический компонент.
Расщепление белка предшественника амилоида может протекать двумя путями — неамилоидным и амилоидным. В первом случае работают α- и γ-секретазы, во втором, как было сказано выше, β- и γ-секретазы. γ-секретаза работает с достаточно большим разбросом — от 30 до 51 аминокислотного остатка, — чем и объясняется вариативность пептида на выходе. После образования Аβ патологический каскад продолжается: данная форма, проходя путь от мономера до амилоидных фибрилл, накапливается во внеклеточной среде, а также вовлекая в процесс как новые Аβ, так и тау-белок по прионному механизму (Рис. 3, 5).
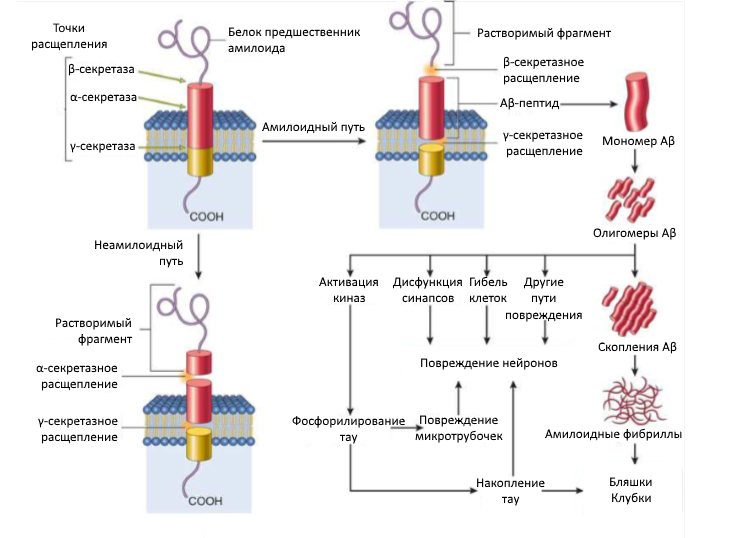
Рисунок 5 | Каскад образования Аβ
Амилоидообразование в рамках ЦНС способно вызвать два заболевания: болезнь Альцгеймера (AD) и церебральную амилоидную ангиопатию (ЦАА).
При AD обнаруживаются два вида патологических включений: амилоидные бляшки и нейрофибриллярные клубки. Последние образованы тау-белком, поэтому рассматриваются ниже.
Амилоидные (сенильные) бляшки — это эллипсоидные нерастворимые образования диаметром 0,1–0,3 мм. Они состоят из внеклеточных скоплений амилоидных фибрилл в центре и дистрофированных отростков нервных клеток по периферии. В случае с бляшками основу составляют Аβ42 и Аβ43. Основные гены, мутации которых доказано вызывают увеличение количества этих форм амилоида, — ген APP (белка предшественника амилоида), пресенилины 1 и 2. И, да, автор знает про ген APOE4 — ведущий генетический фактор риска AD, — но на сегодняшний день нет абсолютных доказательств его связи с изменением соотношения бета-амилоидов.
В своем «развитии» бляшка проходит четыре гистологически различимых этапа: диффузный, примитивный (молодая бляшка), невритический (классическая, или зрелая, бляшка) и этап «выгорания» (Рис. 6). На первом этапе еще не сформировано амилоидное ядро, поэтому бляшка большая, неплотная, слабо окрашивается; с «возрастом» бляшка становится более «концентрированной» — меньше, плотнее, выраженнее.
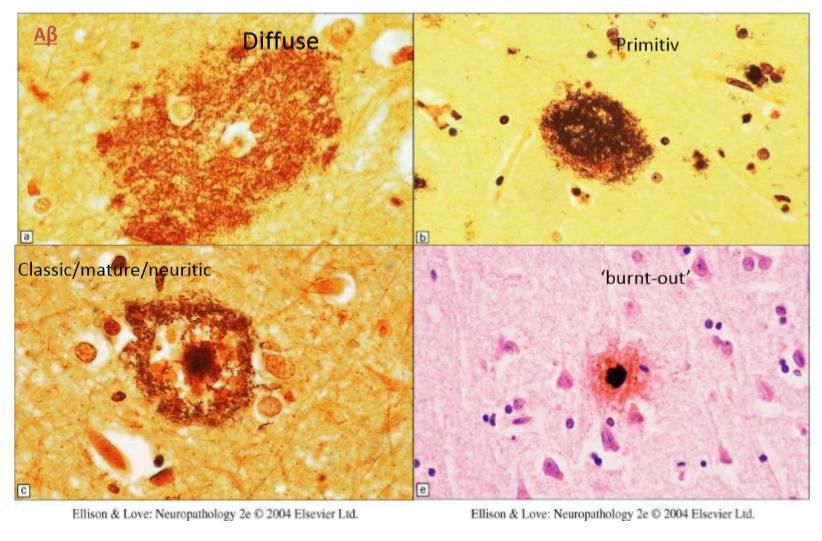
Рисунок 6 | Этапы развития сенильных бляшек
Подобные бляшки также были обнаружены при вскрытии у пациентов с болезнью Крейтцфельдта-Якоба (БКЯ), что подтверждает теорию о непосредственной связи прионных заболеваний и их механизмов «размножения» с другими НДЗ.
А еще бляшки могут быть... заразны. Дело в том, что они практически не растворимы и способны вовлекать другие белки в отряд β-листовых. Это сочетание подразумевает, что при контакте медицинского инструментария с бляшкой часть β-амилоида может остаться на нем, и при недостаточной обработке (недостаточной для его растворения, что немало) возможно, натурально, перенести проблему с больной головы да на здоровую. Теоретически подобный механизм может наблюдаться и у других включений, но на сегодняшний день это остается лишь интересной (и пугающей) теорией.
При ЦАА амилоид, а именно Аβ39 и, реже, 40, откладывается в стенках (конкретно — адвентиции и медии) сосудов головного мозга среднего и малого калибров, прежде всего — кровоснабжающих кору и мозговые оболочки. Концентрация инородных тел в этих слоях стенки обратно пропорциональна эластичности и прочности, что чревато кровоизлияниями (Рис. 7). ЦАА может быть как отдельным заболеванием, так и работать в связке с AD.
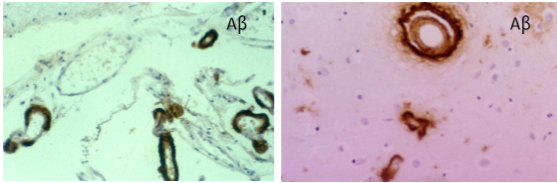
Рисунок 7 | Отложения Аβ39 в сосудистой стенке при ЦАА
На этом первая часть подходит к концу. В продолжении рассмотрим оставшиеся пять видов включений (возможно, четыре, поскольку прионы более чем заслуживают отдельного рассмотрения).
Источники:
- Al-Chalabi A. & HardimanO. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617-628.
- Al-Sarraj S et al: p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72 linked FTLD and MND/ALS. Acta Neuropathologica. 2011; 122:691-702.
- Bettens, K., Sleegers, K. & Van Broeckhoven, C. Genetic insights in Alzheimer's disease. Lancet Neurol. 12, 92–104 (2013).
- Chávez-Gutiérrez, L. et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274 (2012).
- Greenfield's Neuropathology, Ninth Edition, (2015). Eds: Love, Perry, Ironside, Budka CRC Press, ISBN 9781498721288 chapters on ‘Motor neuron disorders’ by Ince P. et al. and ‘Dementia’ by Lowe J. & Kalaria R.
- Hasegawa M. Biochemistry and molecular biology of tauopathies. Neuropathology. 2006; 26:484-90
- IwatsuboT. Pathological biochemistry of α-synucleinopathy. Neuropathology. 2007; 27:474-8
- Keller BA, VolkeningK, et al. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathologica. 2013; 124: 733-747.
- Mackenzie IRA: The role of dipeptide repeat protein pathology in C9orf72 mutation cases.Neuropathol Appl Neurobiol. 2015; ‘on-line first’ DOI: 10.1111/nan.12296
- Neuropathology of Neurodegenerative Diseases — a Practical Guide. First edition (2015) ed.: G.G. Kovacs. Cambridge University Press. ISBN 978-1-107-44242-9. chapter on ‘Amyotrophic lateral sclerosis and frontotemporal dementia’ by Hortobágyi T.& Cairns N.
- Neuropathology. 3rd ed. (2013). Editor: Ellison, Love, et al. Mosby/Elsevier, ISBN 978-0723435150
- Schipper L J. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review.Neuropathol Appl Neurobiol.2015; ‘online first’
- Sreedharan J. et al.: TDP-43 mutations in familial and sporadic ALS. Science. 2008; 319:1668-1672
- Vance C et al.: Mutations in FUS, an RNA processing protein, cause familial ALS type 6. Science. 2009; 323:1208-11.
