
Клеточные технологии в иммунотерапии злокачественных новообразований: химерный рецептор Т-лимфоцитов

Клеточное звено иммунитета является главным оружием организма в борьбе с опухолью. CD8+-лимфоциты, их еще называют Т-киллеры (Тк), способны специфично узнавать антигены на поверхности злокачественных клеток и разрушать их. Для этого на поверхности Тк имеются Т-клеточные рецепторы (T-cell receptor, TCR), похожие на Fab фрагменты иммуноглобулинов, специфичные в отношении определенных опухолевых пептидов, контактное взаимодействие с которыми приводит к активации Тк. Активированный Тк выделяет ряд веществ, таких как перфорины и гранзимы, а также экспрессирует на своей поверхности лиганды клеточной смерти (апоптоза), такие как TRAIL и FasL (рис.1) [1,2].
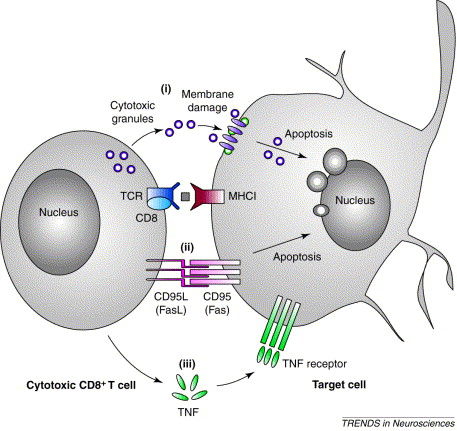
Рисунок 1 | Взаимодействие Тк и опухолевой клетки [2].
Перфорины работают по тому же принципу, что и компоненты системы комплемента — формируют «поры» в мембране клеток, через которые выходит содержимое цитоплазмы (осмотический лизис). Гранзимы относятся к семейству сериновых протеаз и, проникая внутрь «клетки-мишени», активируют механизм апоптоза. Взаимодействие летальных лигандов с рецепторами TRAIL и Fas, которые относятся к суперсемейству фактора некроза опухоли (ФНО), также приводит к запуску программируемой гибели клетки. Кроме того, Тк выделяют ФНО, который запускает апоптоз через соответствующие рецепторы [1].
Однако даже у Тк есть недостатки. Начнем с того, что TCR хоть и обладает высокой специфичностью, но может взаимодействовать только с пептидами и только в составе комплекса гистосовместимости первого класса (Main histocompatibility complex I, MHC-I) [1,3]. Это значит, что CD8+-лимфоциты не реагируют на опухолевые поверхностные углеводы, ганглиозиды и протеогликаны, которые могут обладать большей консервативностью, чем белки. Опухолевые клетки часто перестают экспрессировать MHC-I или извращают процессинг и встраивание белковых антигенов в MHC-I, снижая эффективность TCR [4-6]. Кроме того, Тк несут на своей поверхности рецепторы иммунологических «контрольных точек» PD1 и CTLA4, которые в норме предотвращают гиперактивацию Тк на собственные ткани организма. Многие злокачественные клетки используют этот механизм с помощью лигандов (например, PDL1 и PDL2), предотвращают активацию Тк и вызывают их апоптоз [4,7].
Химерные рецепторы Т-лимфоцитов (Chimeric antigen receptor, CAR) представляют собой искусственно созданные рекомбинантные молекулы, в состав которых входит специфичный антигенраспознающий фрагмент и внутриклеточный участок, передающий сигнал активации Тк (рис. 2) [8].
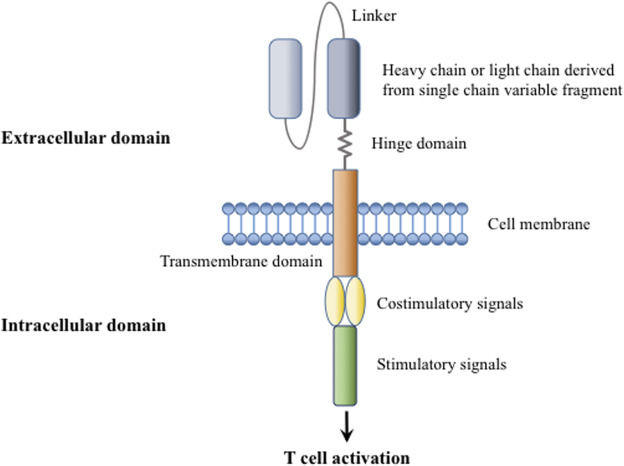
Рисунок 2 | Структура CAR (пояснения в тексте) [9].
Поверхностная часть является антигенсвязывающим фрагментом антитела, а внутренняя — белковыми стимулирующими и костимулирующими молекулами, полученными из TCR. Поскольку части CAR имеют различное происхождение, используется эпитет «химерный» (рис. 3) [8].
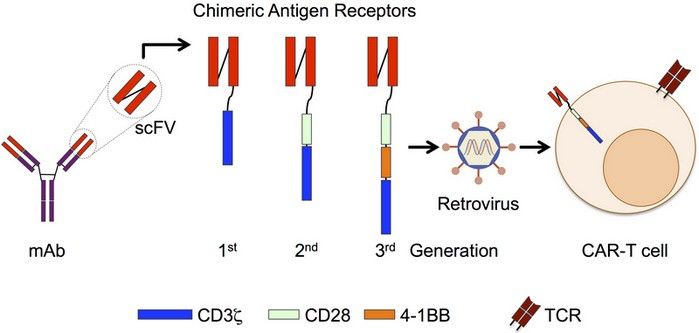
Рисунок 3 | Конструирование CAR и внедрение в Тк (пояснения в тексте) [3].
CAR, в отличие от TCR, способны распознавать любые поверхностные опухолевые антигены без привязки к MHC [10]. Для этой цели используются различные подходы, включая Fab-фрагменты иммуноглобулинов, однако в настоящее время чаще используют одноцепочечный вариабельный фрагмент мышиных антител (single-chain variable fragment, scFV). При этом существует серьезный риск развития иммунной реакции на эти антитела, снижающей эффективность противоопухолевой терапии [8]. Чтобы обойти это ограничение, начались работы по созданию CAR с эндогенным (а значит слабо иммуногенным) антигенраспознающим фрагментом. Такие рецепторы были названы естественные или физиологические CAR (physiological CAR, pCAR). Многие опухоли, например, хронический B-клеточный лейкоз, несут на своей поверхности антиген CD70, который является лигандом рецептора CD27. При этом в нормальных тканях он почти не экспрессируется, за исключением небольших популяций активированных Т- и B-лимфоцитов. Другим рецептором с похожими свойствами является NKG2D, лиганды которого экспрессируются многими гематологическими опухолями, включая лимфомы. Специфичность взаимодействия NKG2D и CD27 используется при создании pCAR, где они используются в качестве антигенраспознающего участка (рис.4) [11,13].
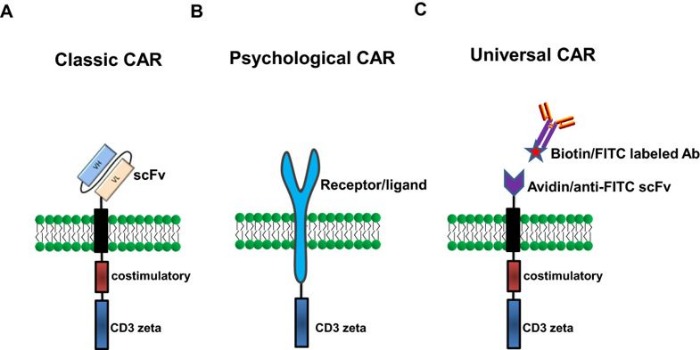
Рисунок 4 | Основные типы CAR. А – классический; B – физиологический (pCAR); С – универсальный [13].
Плюсами такого подхода являются снижение иммуногенности и скорости элиминации Тк с CAR из организма пациента, однако применение pCAR ограничено определенными гистологическими типами злокачественных опухолей [11].
В настоящее время проводятся попытки использования в составе CAR пептидных фрагментов, обладающих антигенраспознающими свойствами, но меньших размеров, чем scFV. В качестве примера можно привести пептидный лиганд T1E, способный связываться с поверхностными рецепторами семейства ErbB. Для той же цели разрабатываются CAR на основе искусственных анкириновых повторов (designed ankyrin repeat proteins, DARPin), наноантител (VHH), а также VLR (variable lymphocyte receptor) [14].
Кроме того, разработка и создание CAR Тк для каждого пациента имеет персональный характер и занимает много времени. Чтобы обойти это ограничение был разработан метод универсальных CAR (universal CAR, uCAR). Общий принцип заключается в создании CAR специфичных к определенным маркерным молекулам. Эти молекулы доставляются к опухоли в комплексе со специфичными антителами, образуя что-то вроде метки на злокачественных клетках, на которую реагируют Тк с uCAR. Примером такого взаимодействия является антигенораспознающий участок — димер авидина курицы, а меткой — антитела с биотином (рис. 4). Кроме того, в настоящее время используются пары: антитела к FITC — флуоресцинизотиоцианат (FITC) и рецептор CD16a — Fc-фрагмент антител [15].
Такие CAR можно использовать против различных типов опухолей и менять молекулярные мишени терапии простым введением меченых антител. Однако авидин и FITC являются достаточно иммуногенными, что затрудняет внедрение этого подхода в практику [15].
Антигенраспознающий участок CAR крепится на шарнирную часть, выбор которой имеет свои нюансы, связанные с правильным подбором расстояния между опухолевой клеткой и Тк в момент взаимодействия CAR с антигеном, но в общем смысле важными является гибкость, длина и иммуногенность этой области, а также способность к трансмембранному транспорту. В большинстве случаев используются следующие молекулы: Fc-фрагменты IgG, CD8α, CD28, CD4, CD7 [11].
Трансмембранный домен служит для закрепления CAR на поверхности клетки, а также влияет на эффективность встраивания CAR в TCR. Для этого используют трансмембранные фрагменты таких молекул, как CD3ζ, CD4, CD8, CD28 [11].
Одной из важнейших частей CAR является внутриклеточный участок, который должен передавать сигнал в цитоплазму Тк и вызывать его активацию. В обычном Тк взаимодействие TCR с MHC-I происходит при костимуляции рецепторами CD8 и CD28. Изменяется конформация TCR и по трансмембранному фрагменту сигнал передается во внутриклеточный домен. У TCR этот участок представлен молекулой CD3, который и вызывает активацию Тк. Первые попытки создать Тк с модифицированным рецептором были неудачными в связи с тем, что при проектировании CAR не учитывался внутриклеточный домен и костимуляторные участки [11].
Первое поколение успешных CAR имело в качестве внутриклеточного домена CD3ζ, как и в обычных TCR. Однако такие рецепторы не могли обеспечить эффективной активации Тк. Связано это с тем, что фосфорилирование CD3ζ активирует каскад киназы ZAP-70, которая является первым сигналом активации Тк. Для полной активации Тк необходимо формирование второго сигнала (каскада PI3K), который запускается костимулирующим рецептором CD28. Говоря простым языком, сигнал 1 приводит к активации Тк и лизису опухолевой клетки, а сигнал 2 к пролиферации Тк (рис. 5) [16].
Чтобы обойти это ограничение, было разработано второе поколение CAR, содержащее в цитоплазматическом участке не только CD3ζ, но и CD28. Иногда в качестве костимулятора используют и другие последовательности, такие как CD134, CD154, CD137, CD27, CD244 и т.д. Второе поколение CAR чаще всего используется в клинической практике (рис. 5) [17].
Кроме этого, разрабатываются CAR, содержащие комбинацию из нескольких костимулирующих последовательностей (третье поколение), а также элементы, приводящие к секреции противоопухолевых цитокинов (четвертое поколение CAR). Активно исследуется возможность создания Тк с модификацией не только антигенсвязывающего рецептора, но и других поверхностных молекул, через которые опухоль может подавлять активность Тк. В качестве примера можно привести Тк, экспрессирующие CD40L, обладающие защитой от опухолевого воздействия и названные за это «армированными лимфоцитами» [11].
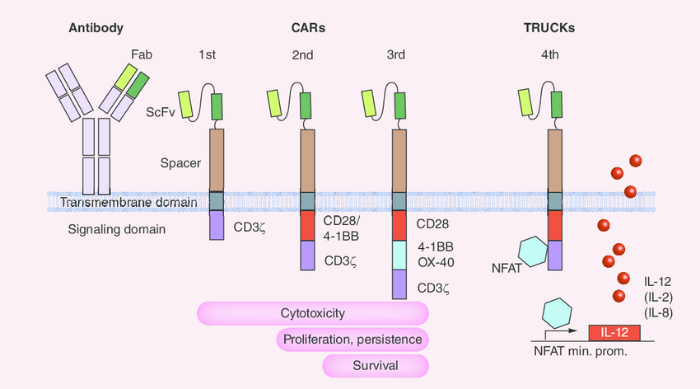
Рисунок 5 | Поколения CAR (пояснения в тексте) [18].
Источники:
- Martínez-lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells?. Clin Cancer Res. 2015;21(22):5047-56.
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25(6):313-9.
- Samstag Y, Emmrich F, Staehelin T. Activation of human T lymphocytes: differential effects of CD3- and CD8-mediated signals. Proc Natl Acad Sci USA. 1988;85(24):9689-93.
- Zhou G, Levitsky H. Towards curative cancer immunotherapy: overcoming posttherapy tumor escape. Clin Dev Immunol. 2012;2012:124187.
- Singh R, Paterson Y. Immunoediting sculpts tumor epitopes during immunotherapy. Cancer Res. 2007;67(5):1887-92.
- Vago L, Perna SK, Zanussi M, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361(5):478-88.
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561.
- Dai H, Wang Y, Lu X, Han W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J Natl Cancer Inst. 2016;108(7)
- Han S, Latchoumanin O, Wu G, et al. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017;390:188-200.
- Almåsbak H, Aarvak T, Vemuri MC. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J Immunol Res. 2016;2016:5474602.
- Liu L, Sun M, Wang Z. Adoptive T-cell therapy of B-cell malignancies: conventional and physiological chimeric antigen receptors. Cancer Lett. 2012;316(1):1-5.
- Magee MS, Snook AE. Challenges to chimeric antigen receptor (CAR)-T cell therapy for cancer. Discov Med. 2014;18(100):265-71.
- Shi H, Sun M, Liu L, Wang Z. Chimeric antigen receptor for adoptive immunotherapy of cancer: latest research and future prospects. Mol Cancer. 2014;13:219.
- Deyev S, Proshkina G, Ryabova A, et al. Synthesis, Characterization, and Selective Delivery of DARPin-Gold Nanoparticle Conjugates to Cancer Cells. Bioconjug Chem. 2017;28(10):2569-2574.
- Salmikangas P, Kinsella N, Chamberlain P. Chimeric Antigen Receptor T-Cells (CAR T-Cells) for Cancer Immunotherapy - Moving Target for Industry?. Pharm Res. 2018;35(8):152.
- Bridgeman JS, Ladell K, Sheard VE, et al. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: implications for optimization of receptor structure for adoptive cell therapy. Clin Exp Immunol. 2014;175(2):258-67.
- Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388-98.
- Kalaitsidou M, Kueberuwa G, Schütt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy. 2015;7(5):487-97.
