
Тяжелая форма наследственной нейтропении (ТФНН, синдром Костманна)
Тяжелые формы наследственной нейтропении включают в себя группу наследственных патологий гемопоэза, которые характеризуются нарушением дифференцировки нейтрофильных гранулоцитов, и, как результат, развивается тяжелая форма хронической нейтропении. При данной патологии абсолютное число нейтрофилов < 0,5 × 109 клеток на литр. Исследование костного мозга у большинства пациентов выявляет подавление миелопоэза на уровне промиелоцитов, что обычно приводит к уменьшению количества нейтрофилов, но в это же время увеличивается количество атипичных промиелоцитов.
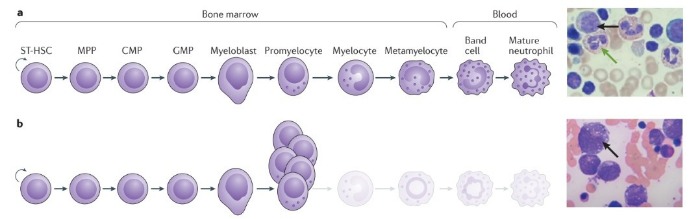
b — У пациентов с ТФНН созревание гранулоцитарных клеток-предшественников останавливается на стадии промиелоцитов и миелоцитов, которые накапливаются в костном мозге. Справа: в мазке костного мозга, окрашенного гематоксилином и эозином, у пациента с тяжелой формой наследственной нейтропении нет зрелых гранулоцитов и увеличено количество промиелоцитов (черная стрелка). Морфология промиелоцитов также различна: у пациентов с ТФНН промиелоциты имеют множественные вакуоли, объемное ядро и они намного больше по размеру, чем промиелоциты у здоровых людей. CMP — общая миелоидная клетка-предшественник; GMP — гранулоцитарно-макрофагальная клетка-предшественник; MPP — полипотентная клетка-предшественник; ST-HSC — гемопоэтическая стволовая клетка.
У этих пациентов риск таких инфекций, как отит, гингивит, кожные инфекции, пневмония, глубокие абсцессы и септицемия, начинает увеличиваться сразу после рождения, и без надлежащего лечения остается высоким на протяжении всей жизни человека. Кроме того, у пациентов с ТФНН повышенный риск развития лейкемии. Мутации в многочисленных генах были связаны с наследственной нейтропенией. Хотя нейтропения является отличительной чертой всех тяжелых форм наследственной нейтропении, тяжесть данного заболевания варьируется и может варьироваться даже у одного и того же пациента с течением времени. ТФНН редкое заболевание: 3-8.5 случаев на миллион человек.
Генетика
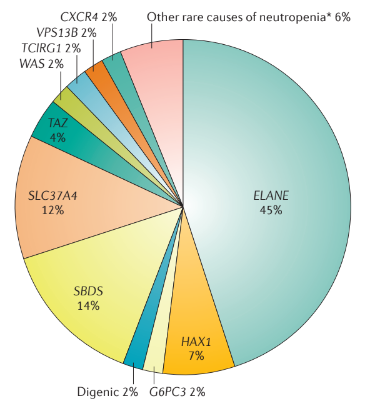
Мутации в гене ELANE. Большинство пациентов с аутосомно-доминантным типом наследования ТФНН являются гетерозиготами и имеют мутации в гене ELANE. Ген ELANE кодирует нейтрофильную эластазу, которая представляет собой цитотоксическую сериновую протеазу, которая хранится в азурофильных гранулах и высвобождается после активации нейтрофилов. Нейтрофильная эластаза гидролизует белковые субстраты, включая белки наружной клеточной мембраны (такие как ГКСФ-рецептор), сосудистый белок клеточной адгезии (СБКА), рецепторы факторов роста тучных и стволовых клеток и рецептор CXC-4 хемокинов. Он также участвует в функции нейтрофильных внутриклеточных ловушек (специфическая сетевидная структура, которая формируется при гибели нейтрофила с высвобождением огромного количества ферментов и фрагментов ДНК. Используется для захвата патогенных микроорганизмов). В данный момент идентифицировано более 200 мутаций гена ELANE, которые случайно распределены по всем экзонам, а также находятся в интроне 3 и интроне 4. При некоторых мутациях гена ELANE, в таких как p.C151Y или p.G214R, более высокий риск развития лейкемии, слабый ответ на терапию посредством ГКСФ (гранулоцитный колониестимулирующий фактор, ГКСФ) и высокий риск развития тяжелых инфекций. К тому же, есть случаи фенотипически здоровых родителей с мозаицизмом мутации гена ELANE (то есть у них есть два типа популяций клеток, один с мутацией гена ELANE, а другой — с ELANE немутантного типа), у которых есть больные дети.
Мутации в гене HAX1. Гомозиготные мутации в гене HAX1 могут быть обнаружены у пациентов с аутосомно-рецессивным типом наследования наследственной нейтропении. Есть две изоформы сплайсинга гена HAX1, и исследования генотипа/фенотипа показали, что мутации HAX1, влияющие на обе изоформы (в основном p.Q190X и p.R86X у японских пациентов) вызывают тяжелые врожденные нейтропении с неврологическими симптомами (например, задержка развития и эпилептические припадки). Мутации, которые влияют только на одну изоформу (в основном p.W44X) приводят к тяжелой форме наследственной нейтропении без неврологических проявлений.
Патогенез
Стресс эндоплазматического ретикулума и апоптоз миелоидных клеток. Мутантная нейтрофильная эластаза не может быть должным образом сформирована, проходить процессинг, секретироваться или растворяться в миелоидных клетках пациентов с ТФНН с мутациями в гене ELANE, в зависимости от того, какой домен (участок полипептидной цепи белка, выполняющий какую-либо его функцию) эластазы был затронут. К тому же, мутация гена ELANE, связанная с нейтропенией, которая нарушает начало трансляции, порождает более короткую форму нейтрофильной эластазы, что приводит к неправильной локализации мутированного белка. Внутриклеточное накопление и изменение расположения мутантной нейтрофильной эластазы индуцирует стресс эндоплазматического ретикулума (ЭР) и активирует реакцию несвернутых белков (РНБ, unfolded protein response), которая ведет к увеличению скорости апоптоза, связанного с активацией шаперона ЭР, регулируемого глюкозой (GRP78). В РНБ участвуют белки клеточной поверхности и секретируемые белки, синтезируемые в эндоплазматическом ретикулуме , где им надлежит свернуться и агрегироваться, прежде чем подвергнуться транспортировке. Изменения в ЭР, препятствующие надлежащему созреванию белков, порождают механизм реакции несвернутых белков. Величина активации РНБ варьируется в зависимости от различных мутаций в гене ELANE. Остается непонятным факт: одни и те же мутации в гене ELANE активируют РНБ у пациентов с ТФНН, но не у пациентов с циклической нейтропенией.
Повышенный апоптоз миелоидных клеток-предшественников в костном мозге наблюдался у пациентов с ТФНН с мутациями в генах ELANE и HAX1. Уменьшение экспрессии антиапоптотических белков (регулятор апоптоза Bcl2 (BCL2), BCL2-ассоциированный агонист гибели клеток (BCLXL) и повтор-содержащий белок бакуловируса IAP (также известный как ингибитор апоптоза сурвивина)) и повышенная экспрессия BCL2-связанного белка A1 (также известный как BFL1) и антиапоптотической изоформы клеточного дифференцировочного белка MCL1, индуцировающего миелоидный лейкоз, были обнаружены при данной патологии.
Дерегулированная экспрессия факторов транскрипции. Помимо нейтропении, у большинства пациентов с ТФНН повышено количество моноцитов и эозинофилов (от двух до четырех раз). Моноцитоз можно объяснить как компенсаторный механизм врожденного иммунитета при отсутствии нейтрофилов. Это также может быть следствием нерегулируемой передачи сигнала транскрипционных факторов, ответственных за коммитирование (приобретение клеткой структурно-функциональных признаков терминальной стадии дифференцировки) миелоидных предшественников до нейтрофилов или моноцитов. У пациентов с ТФНН экспрессия гранулопоэза-активирующего белка CCAAT / энхансер-связывающего белка-α (С/ЭСБ-а) резко снижается, и экспрессия монопоэз-активирующего транскрипционного фактора ПУ.1 (PU.1) не изменяется или немного увеличивается. Таким образом, нарушено С/ЭСБ-а : ПУ.1 соотношение экпрессии с сильным сдвигом к экспрессии ПУ.1. Повышенное количество эозинофилов в крови и в костном мозге также являются типичной чертой у пациентов с ТФНН.
Трансдукция сигнала G‑CSFR. Путь передачи сигнала ГКСФ нарушается у пациентов с ТФНН из-за мутаций в генах, участвующих в ГКСФ передачи сигнала. Это объясняет, почему терапия с очень высокой терапевтической дозой ГКСФ эффективна в большинстве случаев. Пациенты с ТФНН не имеют дефектов в синтезе ГКСФ или в экспрессии ГКСФ-Р (их сыворотка содержит повышенное количество ГКСФ, и миелоидные клетки экспрессируют повышенное количество ГКСФ-Р). Дисрегуляция в трансдукции сигнала ГКСФ-Р происходит на уровне эффекторных белков ниже по ходу его пути, что приводит к серьезному нарушению транскрипции генов, которые способствуют пролиферации или дифференциации миелоидных клеток. Вследствие дифференциация нейтрофилов либо уменьшается, либо прекращается вовсе.
Источники
- Skokowa J. et al. Severe congenital neutropenias //Nature Reviews Disease Primers. – 2017. – Т. 3. – С. 17032.
- Horwitz M. S. et al. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology //Hematology/Oncology Clinics. – 2013. – Т. 27. – №. 1. – С. 19-41.
