
Секвенирование генома. С чего все начиналось?

Предпосылками для формирования генетики как самостоятельной научной области послужило открытие законов Менделя. В дальнейшем в XX веке было сделано четыре открытия, положивших начало развитию генетики [1]:
• установлены клеточные основы наследственности — хромосомы;
• определена молекулярная основа наследственности — двойная спираль ДНК;
• открыта информационная основа наследственности, а также биологический механизм, с помощью которого клетки считывают информацию, содержащуюся в генах;
• изобретены технологии клонирования и секвенирования рекомбинантных ДНК.
Последняя четверть прошлого века была отмечена неустанным стремлением расшифровать сначала гены, а затем и целые геномы [2].
Первая рабочая концепция секвенирования — метод Сэнгера, также известный как метод обрыва цепи, — была предложена в 1977 году. За это открытие Фредерик Сэнгер был удостоен Нобелевской премии по химии в 1980 году. Этот метод секвенирования применялся в течение 40 лет, а его усовершенствование и коммерциализация привели к широкому распространению секвенирования [2].
Описание метода Сэнгера
Секвенирование Сэнгера — метод, при котором используются олигонуклеотидные праймеры для поиска определенных областей ДНК. Этот процесс начинается с деспирализации двухцепочечной ДНК [5]. Одна цепочка ДНК является матрицей для синтеза комплементарной цепочки при помощи фермента ДНК-полимеразы. Реакцию с одной и той же цепочкой проводят в четырех разных пробирках, каждая из которых содержит [3]:
— праймер;
— четыре дезоксинуклеотида (дезоксиаденозинтрифосфат, дезоксигуанозинтрифосфат, дезоксицитидинтрифосфат и тимидинтрифосфат);
— небольшое количество (1 к 100) одного из радиоактивно меченных дезоксинуклеотидов (для визуализации продуктов реакции).
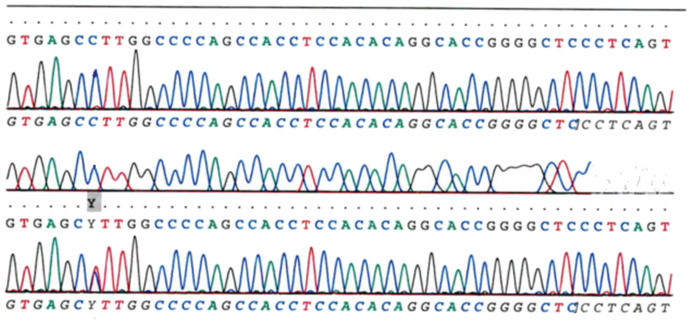
В каждой пробирке образуется набор фрагментов ДНК разной длины, заканчивающихся одним и тем же нуклеотидом. После завершения реакции содержимое пробирок разделяют электрофорезом в полиакриламидном геле в денатурирующих условиях и затем проводят авторадиографию гелей. Каждый дезоксинуклеотид отмечен флуоресцентным маркером: A — зеленый цвет, T — красный, G — черный и C — синий. Лазер в автомате, используемый для считывания последовательности, фиксирует интенсивность флуоресценции. Когда в последовательности встречается гетерозиготный вариант, локусы захватываются двумя флуоресцентными красителями одинаковой интенсивности. Если присутствует гомозиготный вариант, ожидаемый флуоресцентный цвет заменяется цветом комплементарного основания [5].
Продукты четырех реакций формируют «секвенирующую лестницу», которая позволяет «прочитать» нуклеотидную последовательность фрагмента ДНК. Метод Сэнгера позволяет также определять нуклеотидную последовательность РНК, но она предварительно должна быть «переписана» в формат ДНК с помощью обратной транскрипции [3].
Секвенирование Сэнгера — это надежный метод для определения генных мутаций, который широко использовался в течение нескольких десятилетий. Метод Сэнгера является геноспецифичным, и с его помощью анализируют небольшое подмножество генов. Секвенирование Сэнгера позволяет идентифицировать мозаичные мутации. Но метод секвенирования Сэнгера не позволяет проводить точную количественную оценку, то есть нельзя сделать вывод о том, в каком количестве клеток есть мутация [5].
Метод дробовика
Метод дробовика используется для секвенирования длинных участков ДНК. Суть метода состоит в получении случайной массированной выборки клонированных фрагментов ДНК данного организма, на основе которых восстанавливается исходная последовательность ДНК [6].
Первые методы секвенирования способны восстанавливать небольшие последовательности ДНК (порядка 1000 нуклеотидов), следовательно, для секвенирования более длинных последовательностей требовалось разработать новый подход. При секвенировании методом дробовика ДНК случайным образом фрагментируется на мелкие участки с помощью сайт-неспецифичных нуклеаз. Затем фрагменты секвенируют любым доступным методом, например, методом секвенирования по Сэнгеру. Полученные перекрывающиеся случайные фрагменты ДНК собирают с помощью специального программного обеспечения в одну целую последовательность. Данный метод оставался фундаментальным методом секвенирования генома в течение 20 лет [2]. В 1981 году метод применен на практике — полное секвенирование генома вируса мозаики цветной капусты [7].
На практике трудности возникают из-за повторяющихся последовательностей. Например, можно легко секвенировать типичные бактериальные геномы (около 1,5 % повторов) или эухроматическую часть генома мухи (около 3 % повторов). Человеческий геном содержит более чем 50 % повторяющихся последовательностей. Такие особенности усложняют сборку правильной и законченной последовательности генома [2].
В дальнейшем этот подход совершенствовался: были улучшены механизмы фрагментации и клонирования ДНК. В 1990 году был предложен метод секвенирования парных прочтений. Результаты первого применения метода секвенирования парных концов на практике были опубликованы в 1990 году в работе, посвященной секвенированию человеческого гена гипоксантин-гуанинфосфорибозилтрансферазы [4].
При секвенировании парных прочтений ДНК разрезается на случайные фрагменты, которые затем группируются по весу и клонируются в векторах. Клоны секвенируют с обоих концов с использованием метода обрыва цепи, в результате которого образуются две коротких последовательности [4].
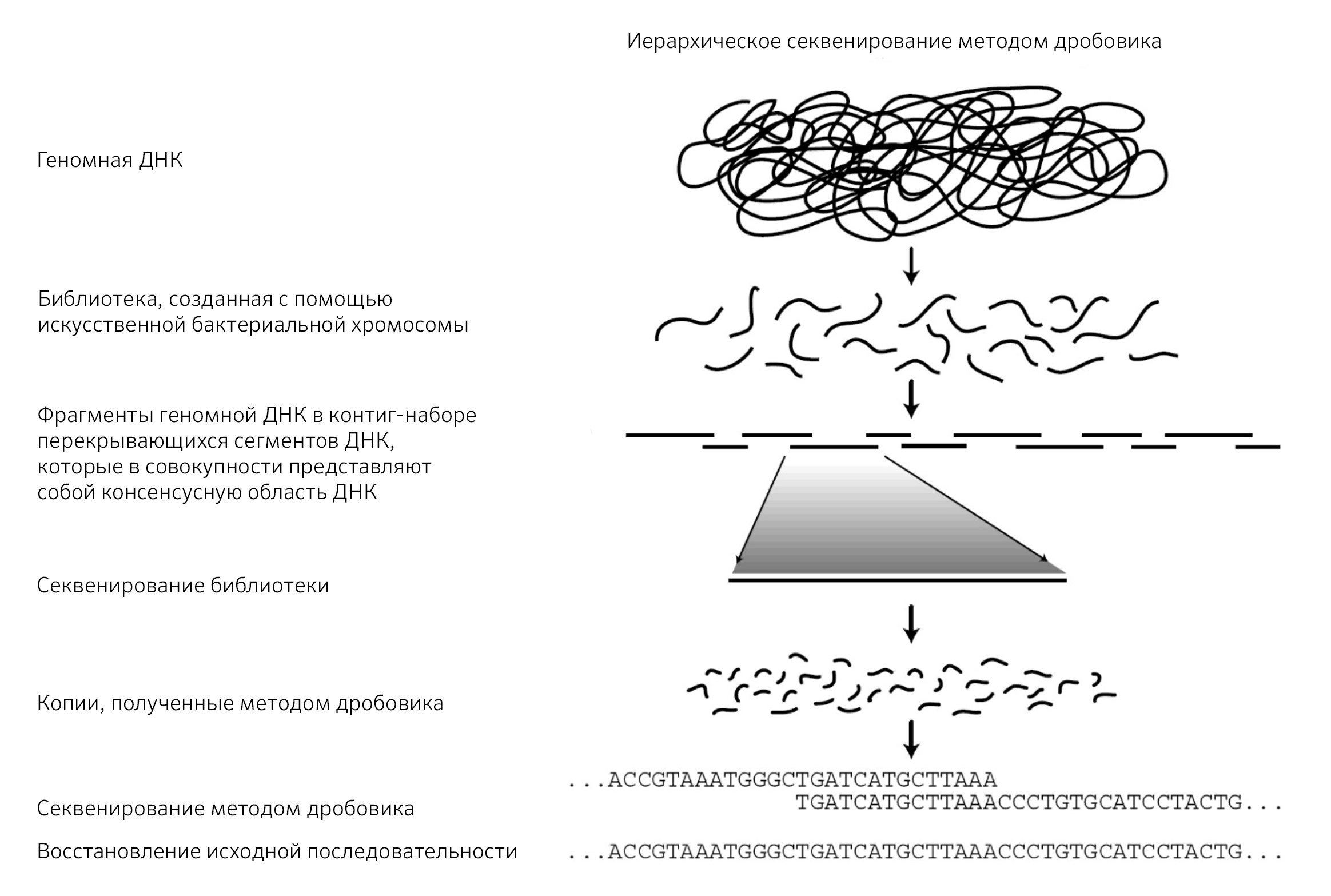
Иерархическое секвенирование методом дробовика
Для секвенирования больших геномов, содержащих повторяющиеся последовательности, используется подход «иерархического секвенирования методом дробовика» [2].
Данный метод — технически более сложный из-за необходимости обработки больших объемов данных. Это служит причиной тому, что метод иерархического секвенирования имеет более высокую стоимость [2].
Переломной точкой развития методов секвенирования стало появление и широкое распространение технологий ПЦР, автоматизация этапов «чтения» ДНК, совершенствование программного обеспечения. Все это дало начало созданию методов секвенирования следующего поколения. Секвенаторы нового поколения значительно дешевле и гораздо эффективнее своих предшественников. На сегодняшний день производительность некоторых секвенаторов измеряется уже сотнями миллиардов пар оснований, что, например, позволяет подобным приборам сканировать индивидуальный геном человека всего за несколько дней.
Источники:
- International Human Genome Sequencing Consortium et al. Initial sequencing and analysis of the human genome //nature. – 2001. — Т. 409. – №. 6822. – С. 860.
- Pareek C. S., Smoczynski R., Tretyn A. Sequencing technologies and genome sequencing //Journal of applied genetics. – 2011. – Т. 52. – №. 4. – С. 413-435.
- Egli M. et al. Nucleic acids in chemistry and biology. – Royal Society of Chemistry, 2006.
- Edwards A. et al. Automated DNA sequencing of the human HPRT locus //Genomics. – 1990. – Т. 6. – №. 4. – С. 593-608.
- Solomon D. A. Integrating Molecular Diagnostics With Surgical Neuropathology //Practical Surgical Neuropathology: A Diagnostic Approach. – Elsevier, 2018. – С. 71-89.
- Staden R. A strategy of DNA sequencing employing computer programs //Nucleic acids research. – 1979. – Т. 6. – №. 7. – С. 2601-2610.
- Gardner R. C. et al. The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing //Nucleic acids research. – 1981. – Т. 9. – №. 12. – С. 2871-2888.
