
Цикл трикарбоновых кислот

Биохимия является основой для глубокого понимания всего, что происходит на более высоких уровнях организации живой материи. Поэтому без опоры на такие знания сегодня немыслимо полноценное биомедицинское образование.
Преподавание курса биохимии будущим врачам гуманной и ветеринарной медицины имеет цель формировать у них универсальные профессиональные компетенции. Предлагаемое учебное пособие продолжает цикл учебных материалов по основам биохимии для студентов и преподавателей факультета «Биоинженерия и ветеринарная медицина» Донского государственного технического университета, а также других вузов медико-биологического профиля. Оно знакомит читателей с процессами жизнедеятельности организма, а также с некоторыми его нарушениями, которые приводят к возникновению болезней, и позволяет, оптимизировать знания в области биохимии, применяя их при изучении нормальной и патологической физиологии, фармакологии, клинической лабораторной диагностики.
ВВЕДЕНИЕ
Автотрофные организмы используют энергию солнечного света для синтеза органических соединений, обладающих запасом внутренней энергии, из СО2 и H2O, то есть получают углерод из СО2. В свою очередь, гетеротрофные организмы используют энергию органических соединений, поступающих с пищей, то есть получают углерод из органических соединений. Основным способом преобразования этой энергии для нужд жизнедеятельности является биологическое окисление или тканевое дыхание. Под тканевым дыханием понимается катаболизм органических веществ клетками с участием O2 и выделением СО2 и H2O. Больше всех от обогащения атмосферы Земли кислородом во времена протерозоя выиграли альфа-протеобактерии.
Концентрация O2 в различных клетках колеблется от 6 до 175 мкМ, и основная его часть, потребляемая эукариотами, восстанавливается в митохондриях до H2O. Любопытно, что кислород является сильным окислителем, но кинетически представляет собой довольно инертную молекулу, и способность к ее ферментативному восстановлению в клетке путем поочередного присоединения электронов имеют металлы переменной валентности (Fe2+/Fe3+, Cu+/Cu2+, Mn2+/Mn3+), а также низкомолекулярные органические соединения (хиноны, флавины, которые являются кофакторами и коферментами соответственно). Общий путь катаболизма является эволюционно сложившимся метаболическим путём. Этот метаболический путь состоит из трех этапов:
- окислительное декарбоксилирование пировиноградной кислоты (пирувата) до ацетил-КоА;
- цикл трикарбоновых кислот (ЦТК) в митохондриях (хотя бывают исключения, например, ГАМК-шунт в нейронах, у растений);
- создание протонного градиента при помощи Цепи переноса электронов (ЦПЭ) или электрон-транспортной цепи.
Все три этапа связаны: конечный продукт одного является начальным субстратом следующего.
ЦТК — филогенетически старый метаболический путь, но не самый древний. Но большая часть организмов, в том числе прокариоты, имеют те или иные компоненты ЦТК. Вообще, синтез, взаимопревращение и разрушение промежуточных метаболитов (пируват, ацетил-КоА), а также все компоненты ЦТК считаются приобретением эукариот вследствие их симбиогенеза с прокариотами (альфа-протеобактериями). Последние в дальнейшем преобразовались в эукариотические органеллы — митохондрии [1]. Такая теория находит отклик в том, что ферменты ЦТК синтезируются с использованием не только митохондриальных генов, но и ядерных.
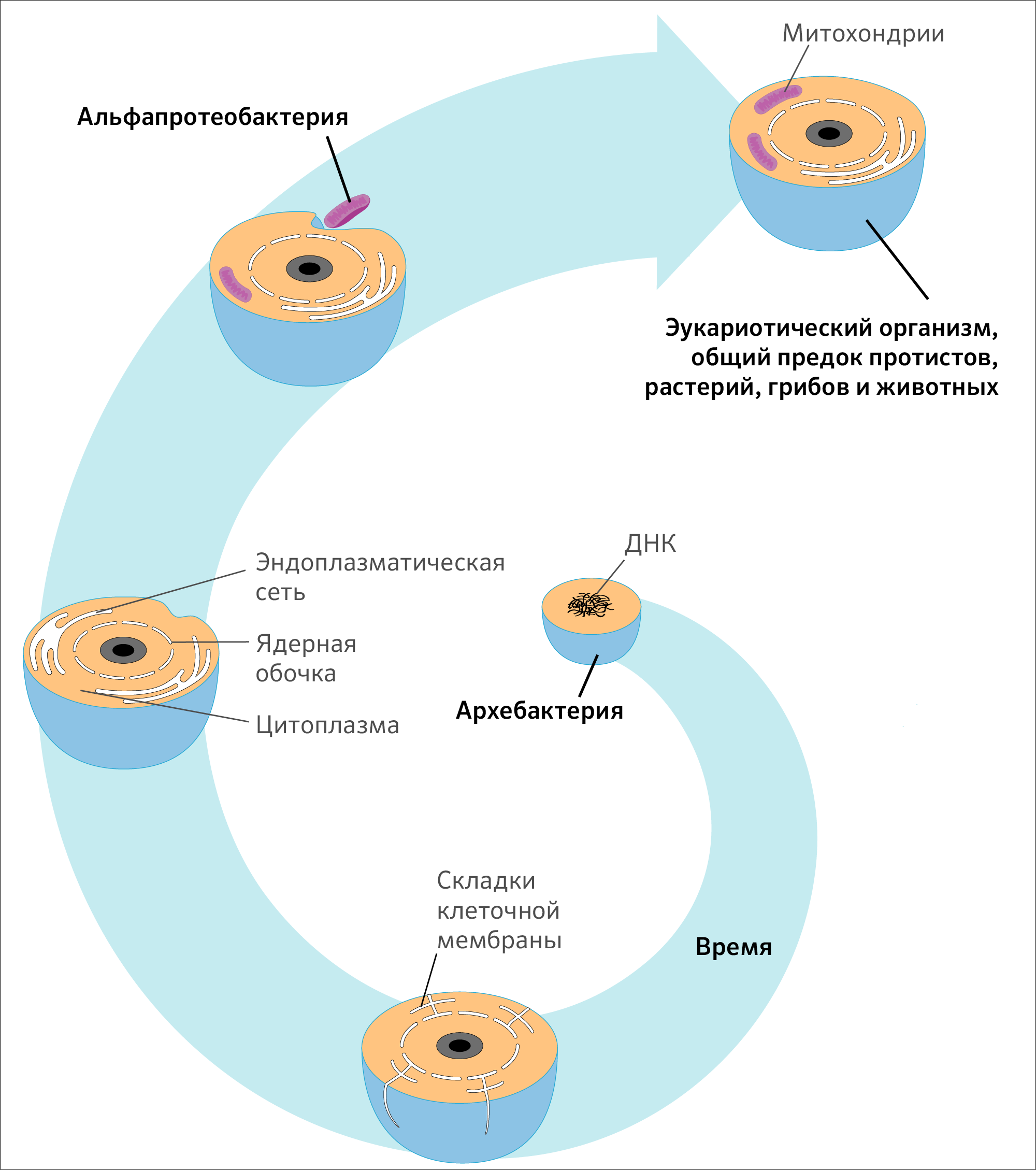
Примечательно, что ЦТК не имеет прямого отношения к аэробному дыханию (филогенетически более новому способу окисления по сравнению с анаэробным дыханием), но снижение концентрации О2 у аэробов подавляет его реакционную способность. Дело в том, что синтезированные в ходе ЦТК НАДH+Н+ далее переносят гидрид-анионы на ЦПЭ и восстанавливают кислород, поступивший в процессе дыхания, до H2O. Образуемый потенциал между митохондриальным матриксом и межмембранным пространством превращается в энергию связей АТФ.
НАД (никотинамидадениндинуклеотид) является коферментом и активированным вариантом никотиновой кислоты (ниацин, витамин В3). Ферменты, обеспечивающие синтез НАД+ из никотиновой кислоты, сконцентрированы в ядре клетки.
Преобразования веществ в ЦТК носят окислительный характер. На сегодняшний день исследовано множество анаэробных организмов, у которых есть набор метаболитов ЦТК, но вместо последовательного окисления субстраты подвергаются восстановлению [2]. Такой цикл получил название восстановительный цикл трикарбоновых кислот (ВЦТК) или цикл Арнона.
Таким образом, компоненты ЦТК (рисунок 2) и ВЦТК (рисунок 3) являются универсальной последовательностью для известных форм жизни [3].
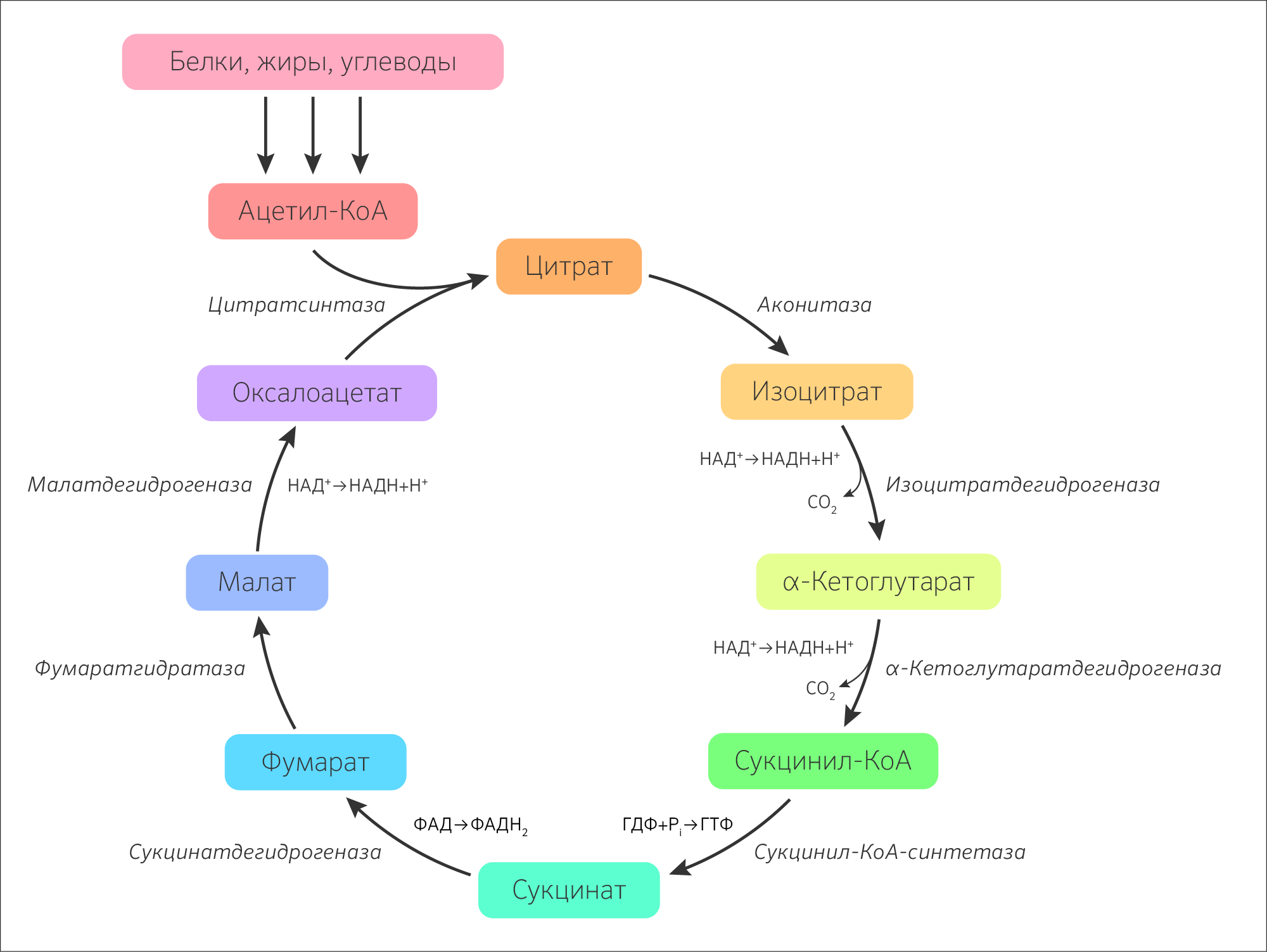
Рисунок 2. Реакции цикла трикарбоновых кислот (цикл Кребса)
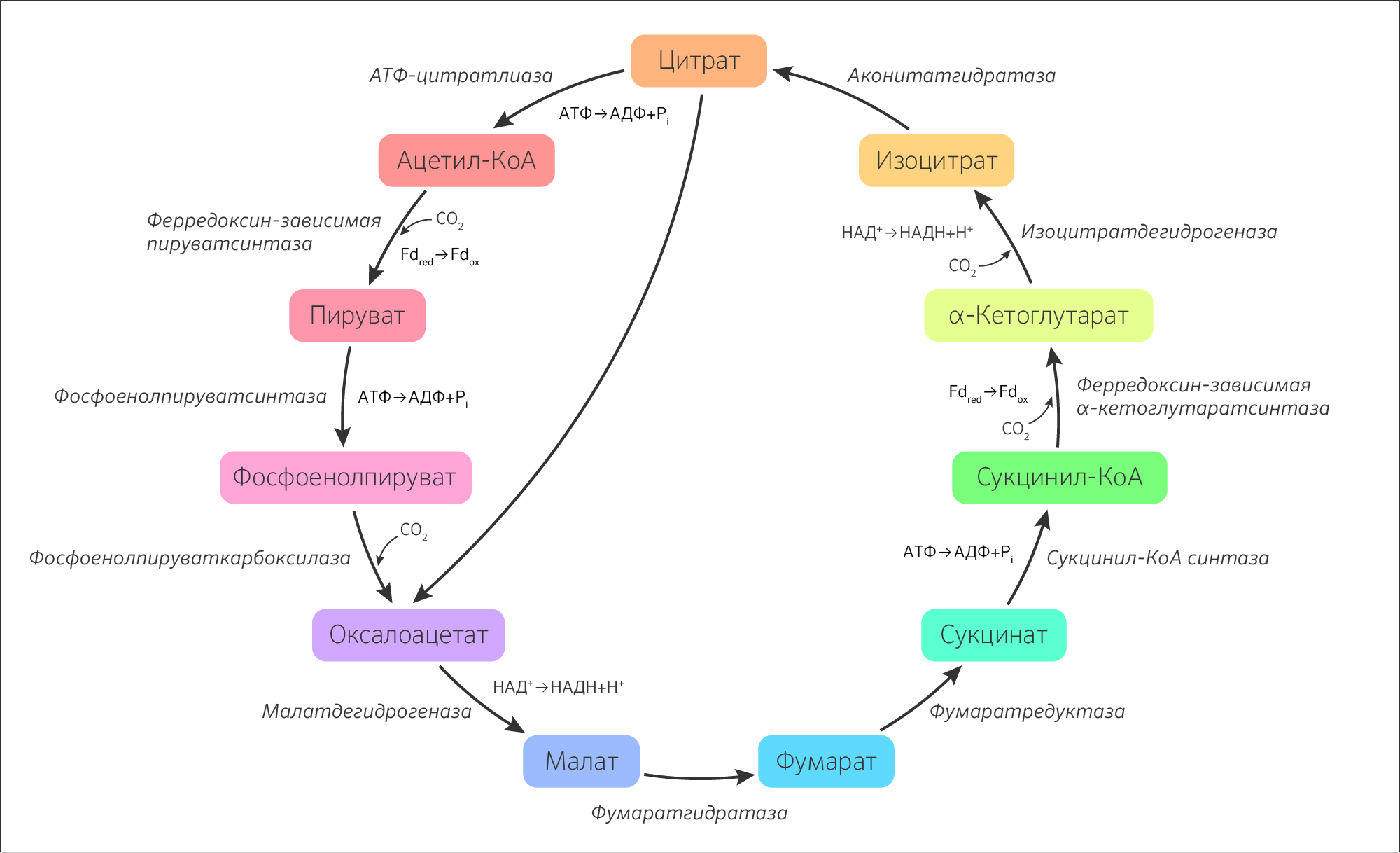
Рисунок 3. Восстановительный цикл трикарбоновых кислот (цикл Арнона)
Хемотрофы (водородобактерии, метанобактерии, железобактерии) — прокариоты и, пожалуй, одни из самых древних форм жизни, начало которым было положено в архей (отсюда и одноименное название домена — Archaea). Для них именно ВЦТК является началом путей биосинтеза органических веществ. Например, хемолитоавтотрофы рода Thiobacillus получают энергию в результате окислительно-восстановительных преобразований неорганических соединений и в процессе хемоассимиляции СO2. В настоящее время нет достоверных сведений о наличии ВЦТК у эукариот [4,5], поэтому для изучения метаболических путей взаимопревращения углеродсодержащих веществ логично рассмотрение не варианта ВЦТК, в котором имелся какой-то примитивный источник энергии, не представленный органическими веществами, а ЦТК как более позднего и нового приобретения эволюции.
ИСТОРИЯ ОТКРЫТИЯ ЦТК
В 1930 году некоторые из компонентов ЦТК открыл Альберт Сент-Дьерди. В частности, он установил, что при добавке сукцината, фумарата и малата к измельченной мышечной ткани поглощается большее количество кислорода, чем требуется для окисления, тем самым придя к выводу, что кислоты являлись катализаторами, но сами не претерпевали изменений. Сент-Дьерди первый, кто непосредственно описал ЦТК и в 1937 году был удостоен Нобелевской премии по физиологии и медицине «За исследования биологического окисления и в особенности за открытие витамина С и катализа фумаровой кислотой» [6].
Однако полную последовательность реакций и образующихся соединений в 1937 году установил Ханс Адольф Кребс. В 1953 году он получил Нобелевскую премию «За открытие цикла лимонной кислоты» (разделил с Фрицем Альбертом Липманом, получившим премию «За открытие кофермента А и его значения для промежуточных стадий метаболизма») [7]. С тех пор ЦТК имеет авторское узнаваемое название — цикл Кребса.
Для чего нам эта историческая справка? Что ж, кто-то разбрасывает камни, а кто-то умело их собирает… Если рассматривать ее в контексте медицины, то эти прорывные, сложные и даже революционные открытия были сделаны не так уж и давно. Мы знаем мало. Мы в начале пути.
ЦИКЛ КРЕБСА
Пусковым субстратом ЦТК является ацетил-КоА. Разберем, из чего он может синтезироваться.
Углеводы
Возьмем всем известный моносахарид — глюкозу, — которая в организме человека может подвергаться гликолизу (рисунок 4). В результате гликолиза из одной молекулы глюкозы получается две молекулы пирувата. Суммарное уравнение гликолиза будет выглядеть так:
Глюкоза + 2НАД+ + 2АДФ + 2Н3РО4 → 2Пирувата + 2НАДH + 2Н+ + 2ATФ + 2Н2O
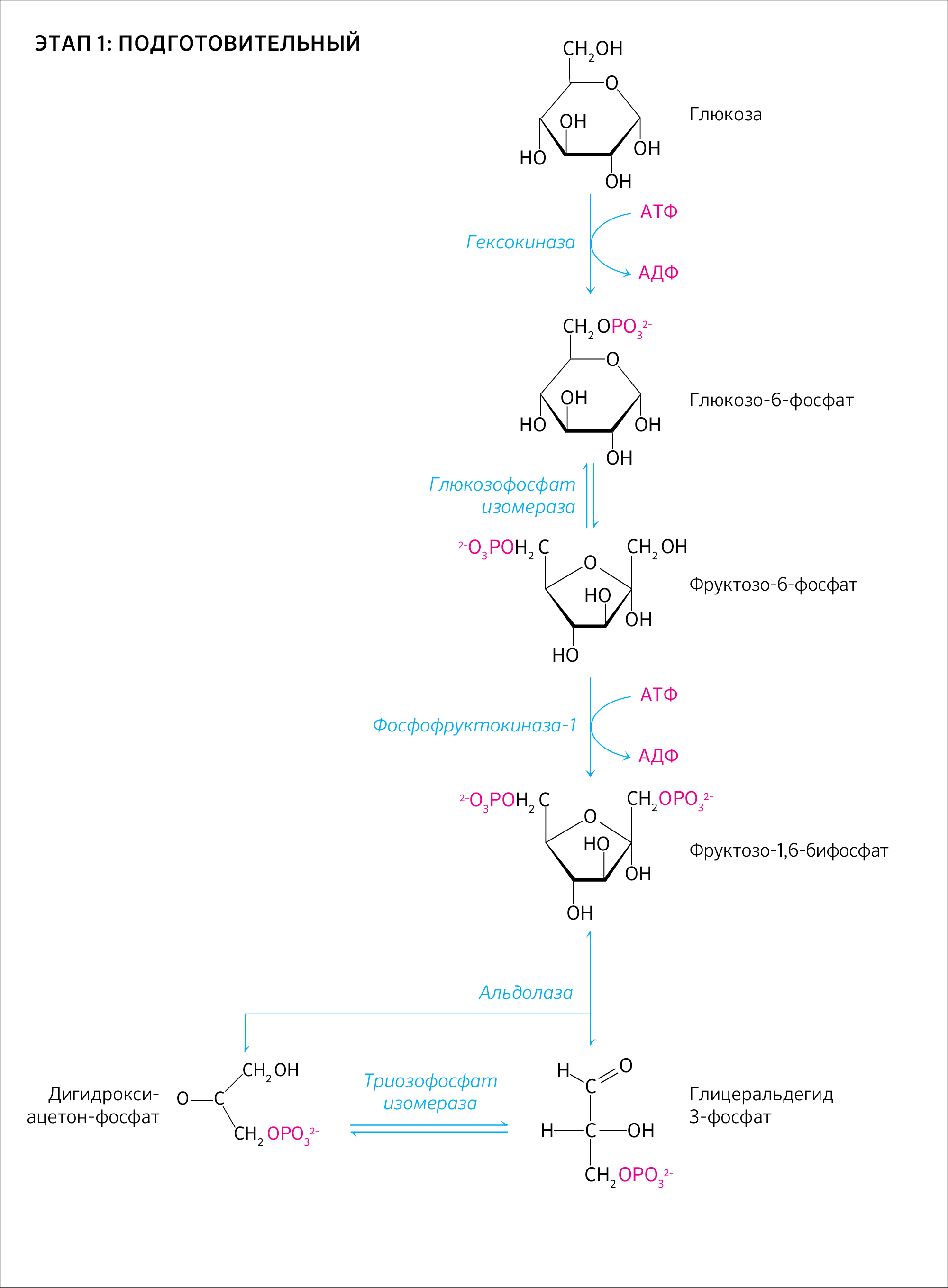
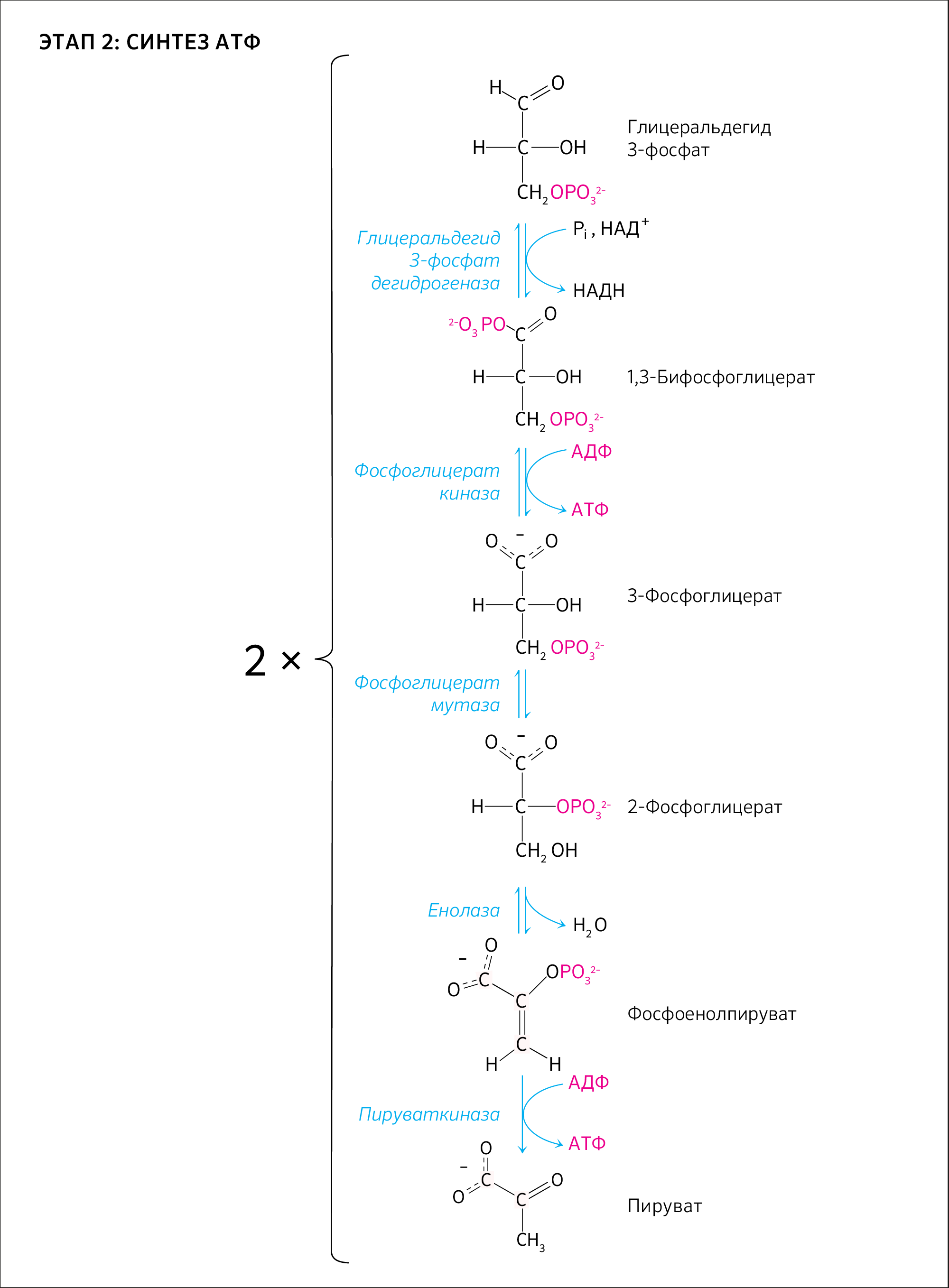
Стоит оговорить некоторые детали. Гликолиз — это не только окисление глюкозы. Другие углеводы тоже окисляются по этому пути, но только в том случае, когда они превращаются в его компоненты. Например, фруктоза при участии одной молекулы АТФ превращается в фруктоза-6-фосфат, и такое преобразование происходит в мышцах и почках. Гликолиз — это анаэробный процесс. Так, в эритроцитах он является единственным путем поддержания их биоэнергетики.
Синтез пирувата осуществляется в цитоплазме клеток в результате гликолиза, а процесс ЦТК идет в матриксе митохондрий. Следовательно, пирувату надо попасть из одного компартмента в другой. Проблема в том, что пируват является полярной молекулой и не может проникнуть через внутреннюю мембрану митохондрии, проницаемую только для О2, СО2 с помощью простой диффузии, в то время как внешняя мембрана проницаема для малых молекул и ионов за счёт поринов.
Транспорт пирувата в митохондриальный матрикс происходит при участии белка транспортера, который по последней классификации IUPAC (Международного союза теоретической и прикладной химии) и NC-IUBMB (Комитета по номенклатуре Международного союза биохимии и молекулярной биологии) относят к транслоказам с механизмом симпорта H+ во внутренней мембране. Для участия в ЦТК необходима ацетильная группа пирувата — СН3–С=О, — однако этому мешает полярная карбоксильная группа, отщепить которую возможно путем окислительного декарбоксилирования с участием специального мультиферментного комплекса в митохондриальном матриксе — пируватдегидрогеназного комплекса. В итоге образуется необходимый ацетил-КоА, и реакция становится необратимой (рисунок 5).
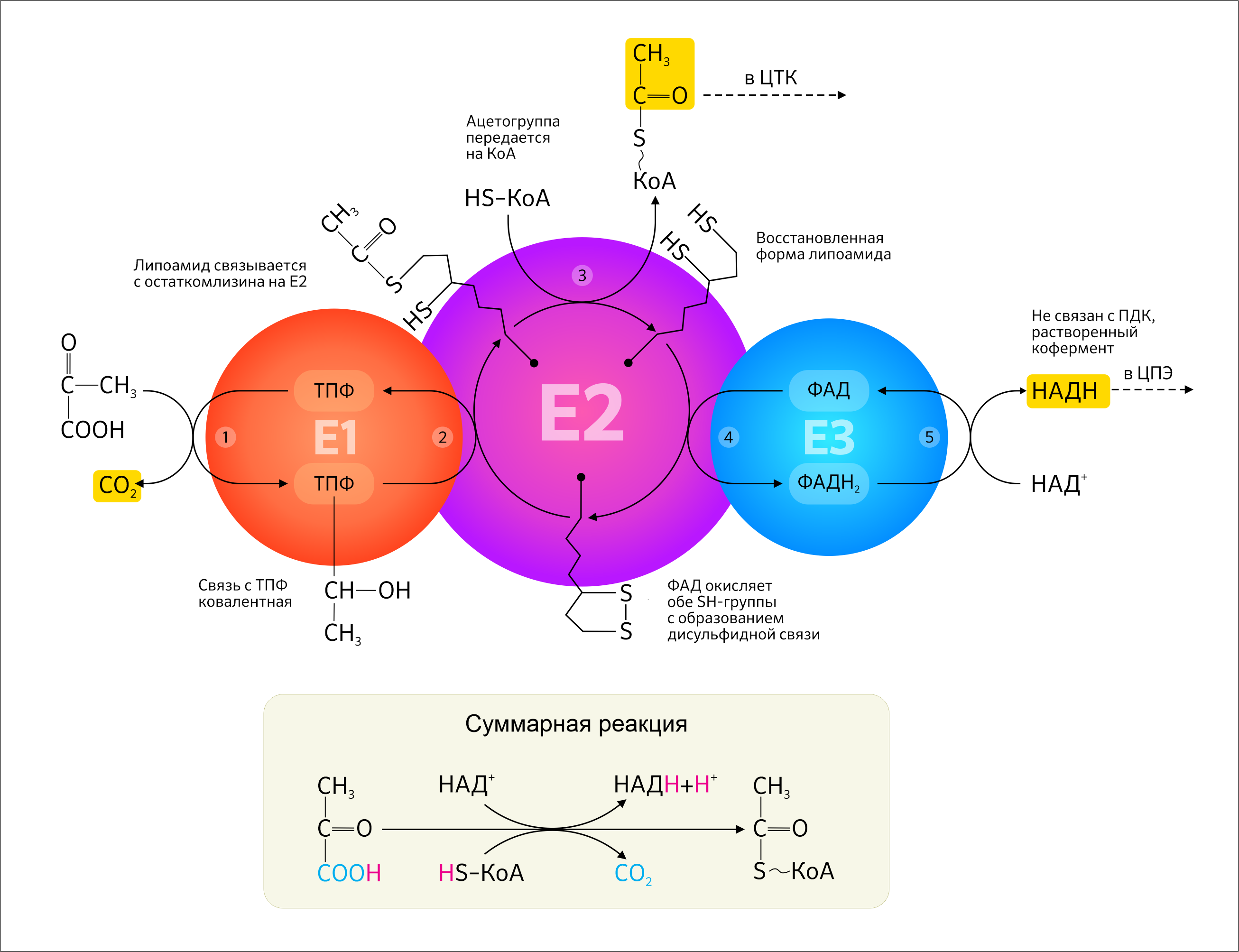
II — Е2 катализирует перенос ацетильного остатка с липоевой кислоты на свободный КоА;
III — ацетилированная дигидролипоилтрансацетилаза взаимодействует с SH группой КоА с образованием восстановленной формы липоевой кислоты (дитиоловая форма) и ацетил-КоА;
IV — окисленная форма трансацетилазы регенерируется при участии Е3, которая обеспечивает перенос двух гидрид-анионов от восстановленных липоильных групп Е2 на кофермент ФАД фермента Е3;
V — окисленная форма Е3 регенерируется. Образованный в Е3 ФАДH2 переносит ионы H+ на НАД+ с образованием НАДН+H+.
Пируватдегидрогеназный комплекс включает в себя три типа ферментов в множестве копий, пять коферментов, два вспомогательных белка.
Ферменты:
- пируватдегидрогеназа (Е1);
- дигидролипоилтрансацетилаза (Е2);
- дигидролипоилдегидрогеназа (Е3).
Коферменты:
- тиаминпирофосфат;
- ФАД;
- КоА;
- НАД+;
- липоат.
Среди компонентов этих коферментов есть четыре витамина и одно витаминоподобное вещество, которые человек получает преимущественно с пищей:
- тиамин (B1) для синтеза тиаминпирофосфата;
- рибофлавин (B2) для синтеза ФАД;
- пантотенат (B5) для синтеза КоА;
- ниацин (B3) для синтеза НАД;
- липоевая кислота (липоат).
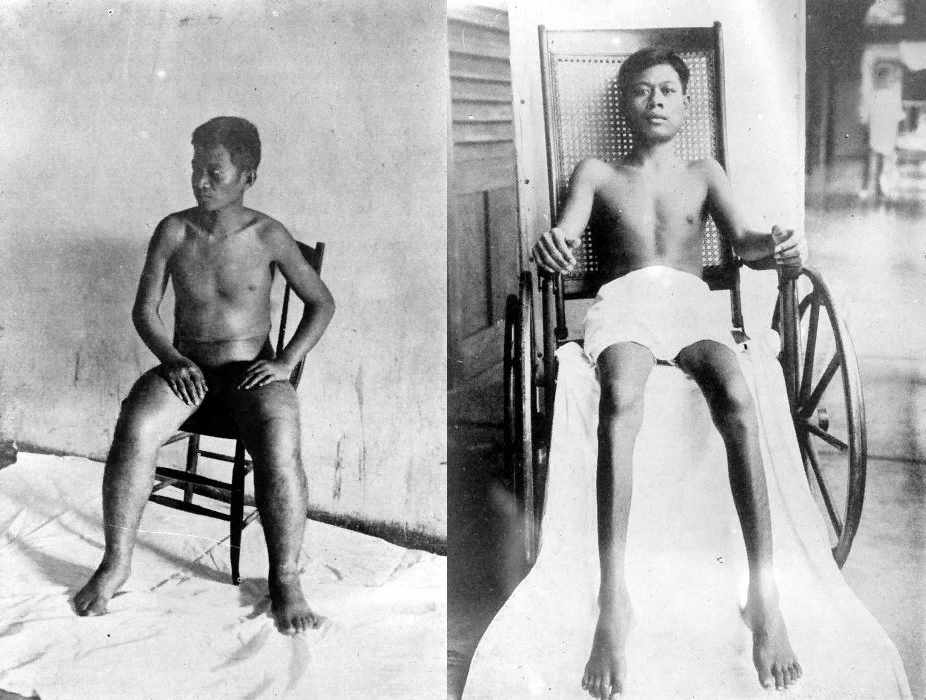
Кстати, далее, по ходу всех стадий, будет понятна роль витаминов в ЦТК. Стоит обратить внимание, что при алиментарной недостаточности компонента пируватдегидрогеназного комплекса тиамина (витамин В1) развивается болезнь бери-бери (рисунок 6), которая на сегодняшний день встречается редко и характерна для слаборазвитых стран.
Вспомогательные белки для протекания сопутствующих реакций фосфорилирования:
- протеинкиназа;
- фосфопротеинфосфосфатаза.
Липиды
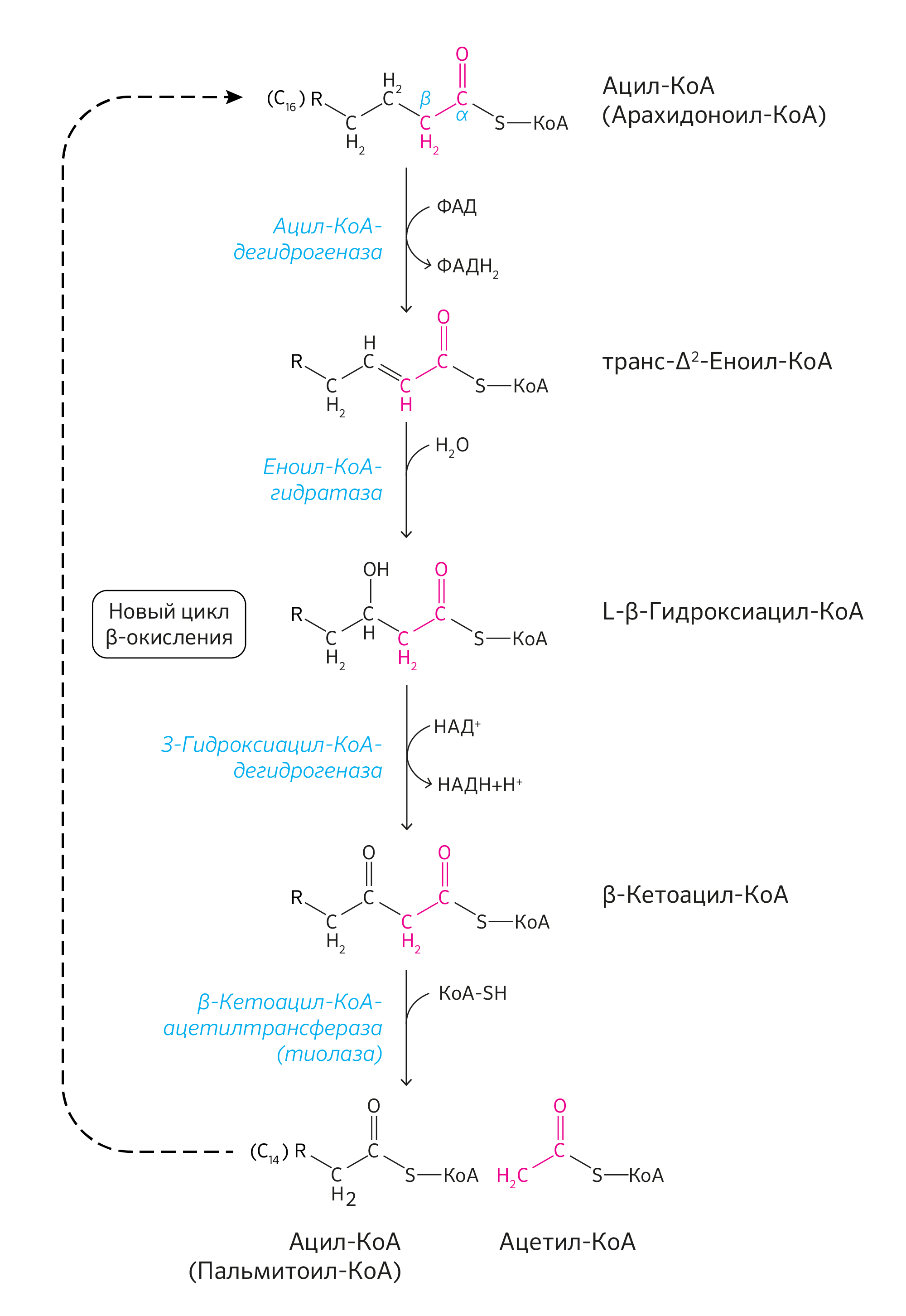
Вспомним липиды, которые распадаются до высших жирных кислот (ВЖК) и глицерола. ВЖК затем подвергаются процессу β-окисления до ацетил-КоА (рисунок 7). Глицерол в свою очередь является субстратом для глюконеогенеза в печени, и уже не сложно догадаться, что будет с вновь синтезированной глюкозой дальше.
Белки
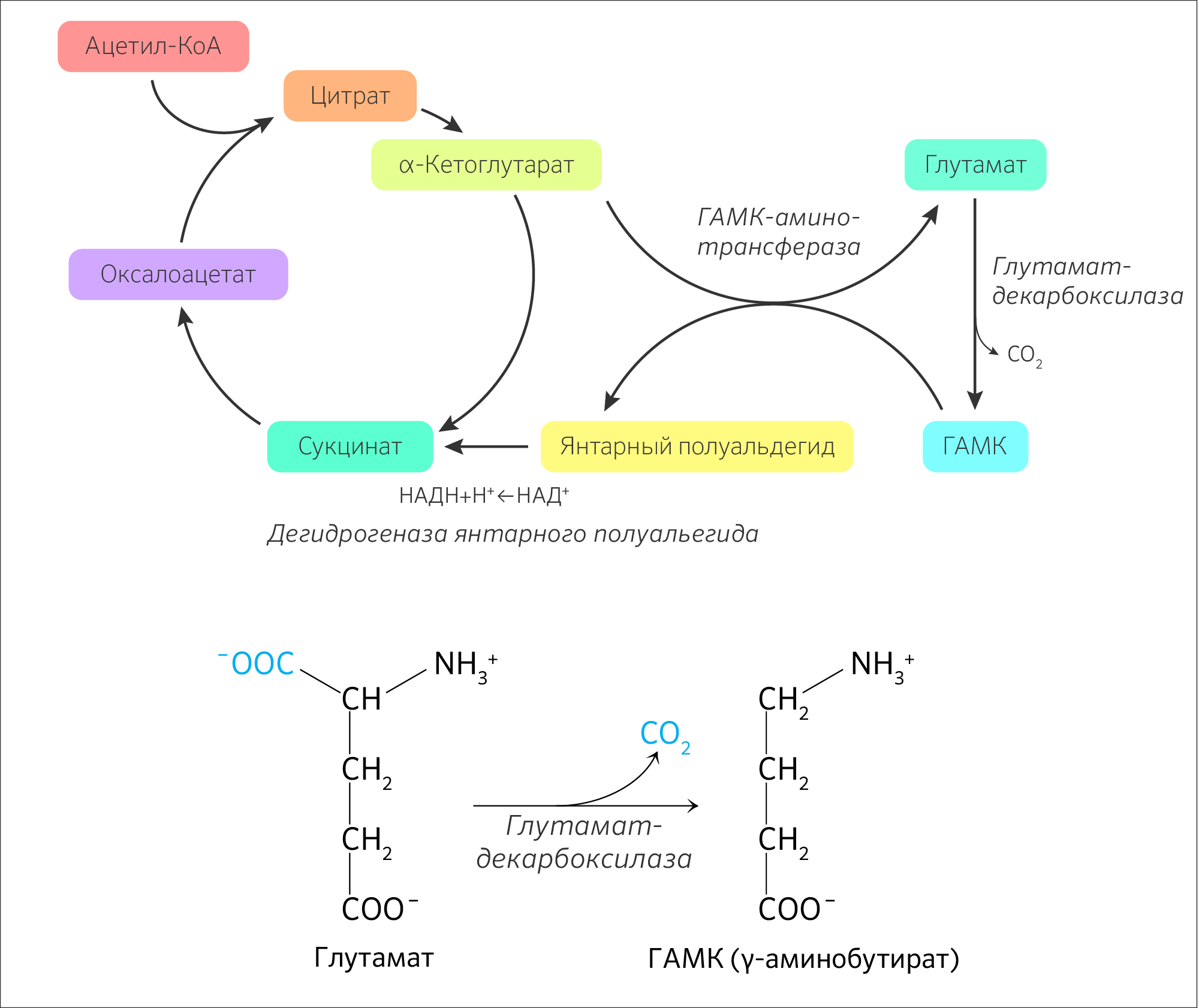
Они очевидно распадаются до аминокислот. И самое важное, что стоит отметить: аминокислоты невероятно пластичны. Путем реакций трансаминирования (ферментативные реакции переноса аминогруппы с аминокислот на кетокислоты) (рисунок 8) организм может получать не только глюкозу и пируват, но также субстраты ЦТК — α-кетоглутарат, оксалоацетат. Более того, такие аминокислоты, как серин, аланин, триптофан и другие могут превратиться в ацетил-КоА, минуя превращение до пирувата (рисунок 9).
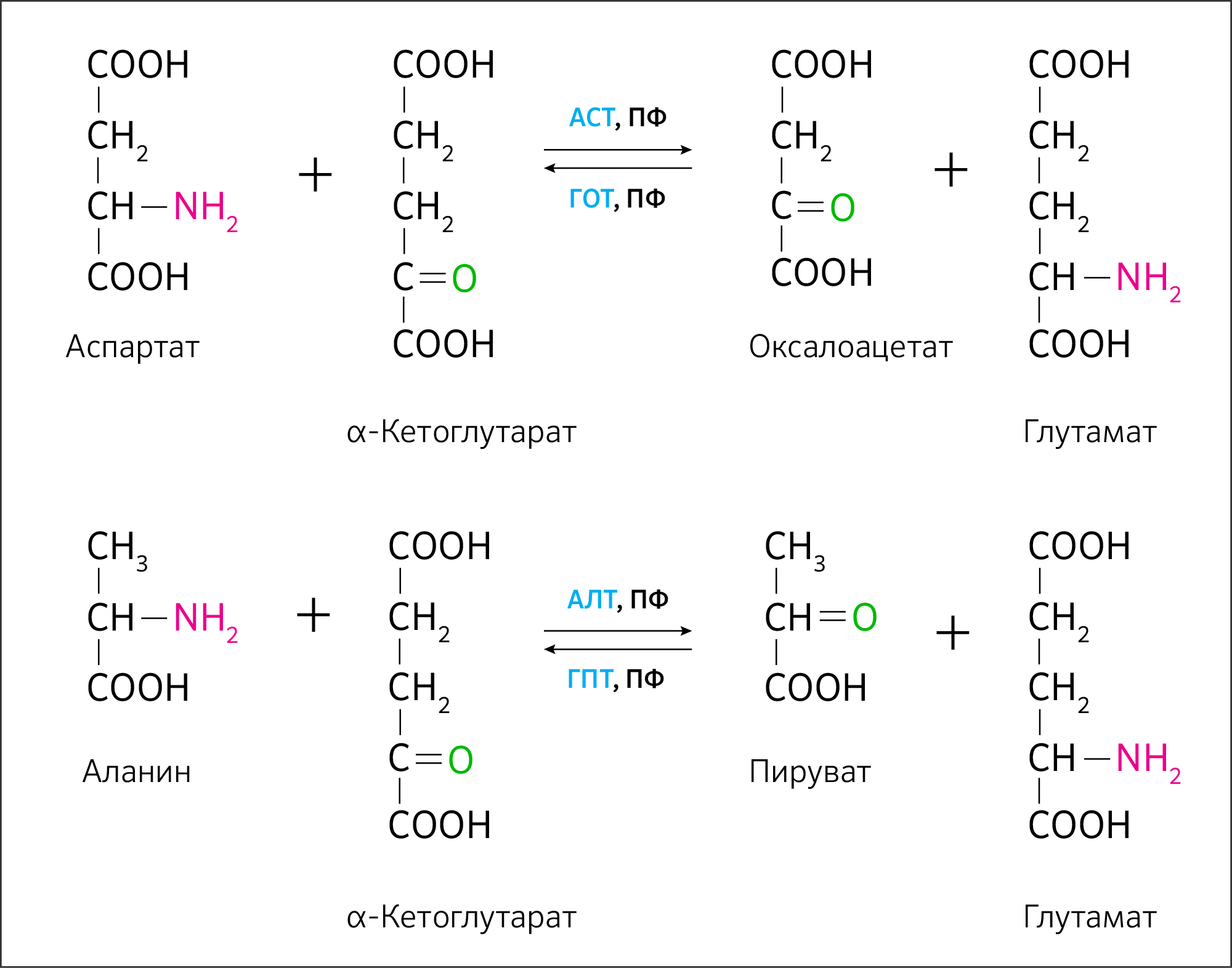
АСТ — аспартатаминотрансфераза, ПФ — кофермент пиридоксальфосфат (производный витамина В6), ГОТ — глутамат-оксалоацетатаминотрансфераза, АЛТ — аланинаминотрансфераза, ГПТ — глутамат-пируватаминотрансфераза
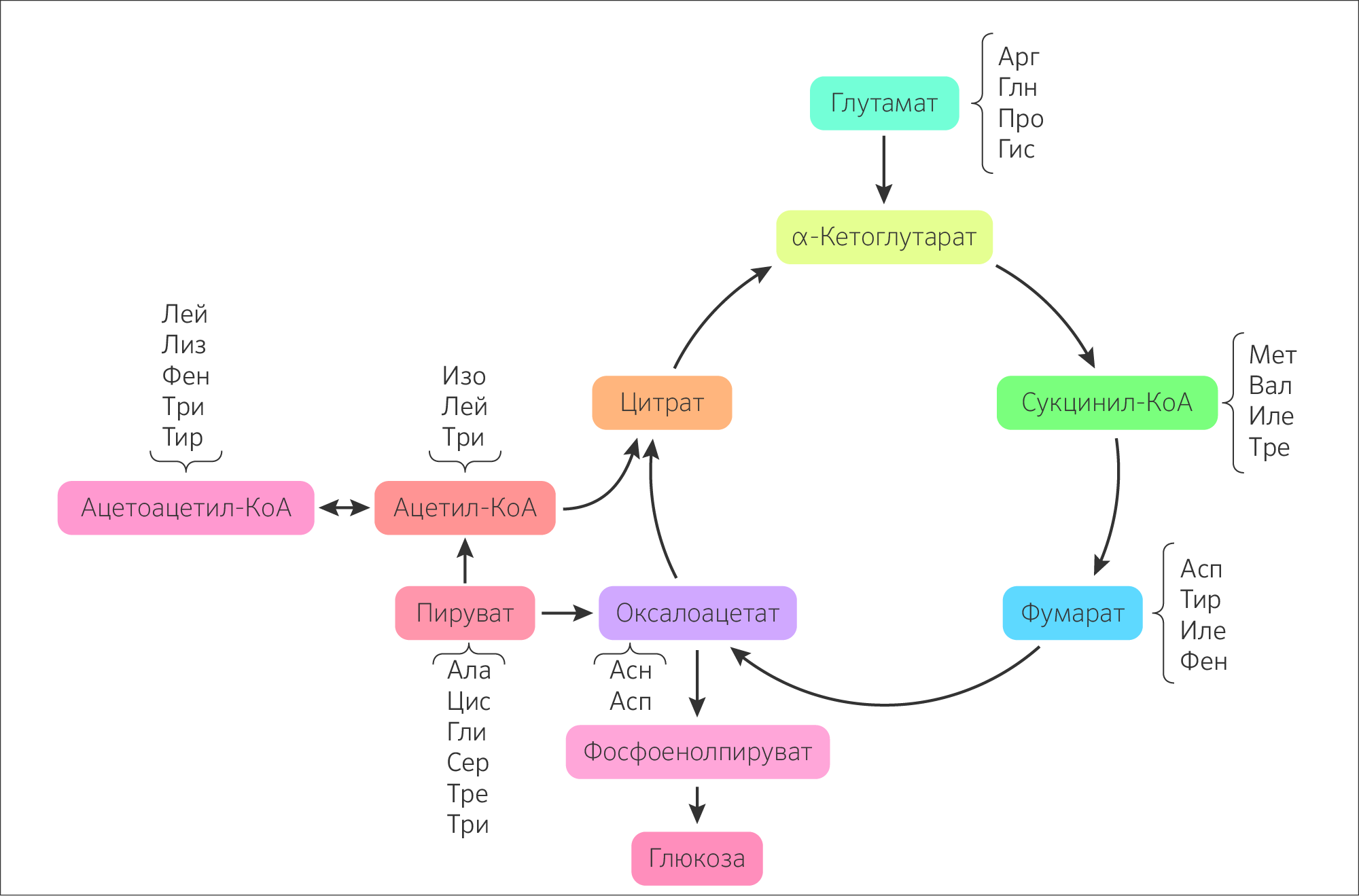
Рисунок 9. Аминокислоты как ресурс для синтеза компонентов цикла трикарбоновых кислот.
Этанол
Алкогольдегидрогеназа печени окисляет этанол до ацетальдегида (рисунок 10). В ходе последующей реакции токсичный ацетальдегид в той же печени при участии альдегиддегидрогеназы окисляется до уксусной кислоты, которая претерпевает превращения с образованием ацетил-КоА.
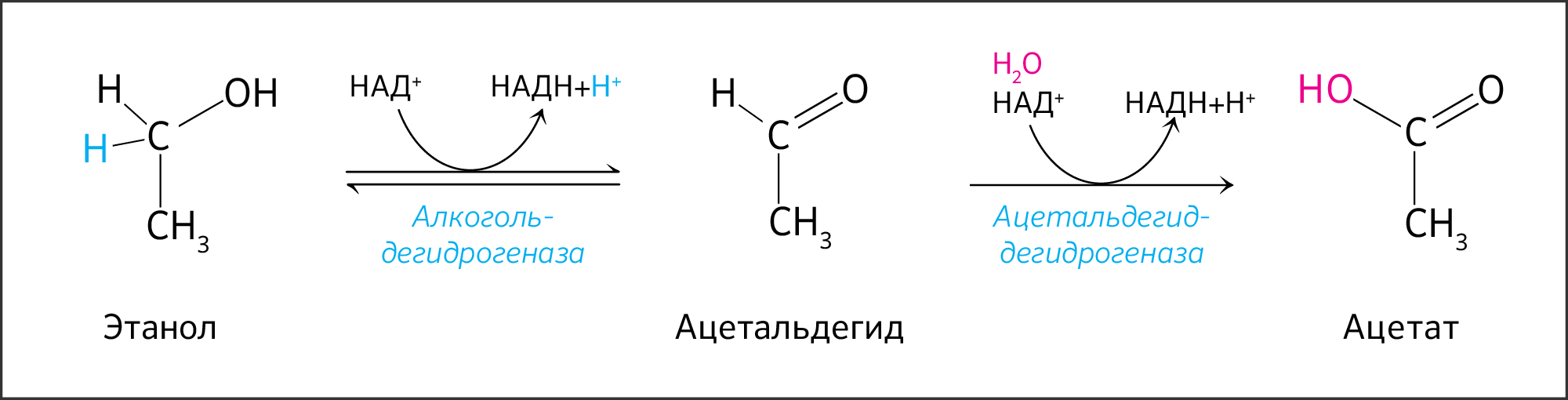
Рисунок 10. Реакция превращения этанола в ацетат. Алкогольдегидрогеназа использует НАД+ для осуществления реакции и в норме не локализуется в сыворотке крови человека.
Кетоновые тела
Если для синтеза ацетоацетата (рисунок 11) в митохондриях гепатоцитов необходимо наличие двух молекул ацетил-КоА, пришедших в результате обильного поступления ВЖК в печень или при обезвреживании этанола, то его катаболизм клетками организма (рисунок 12) вновь приводит к образованию двух молекул ацетил-КоА, которые отправятся в ЦТК.
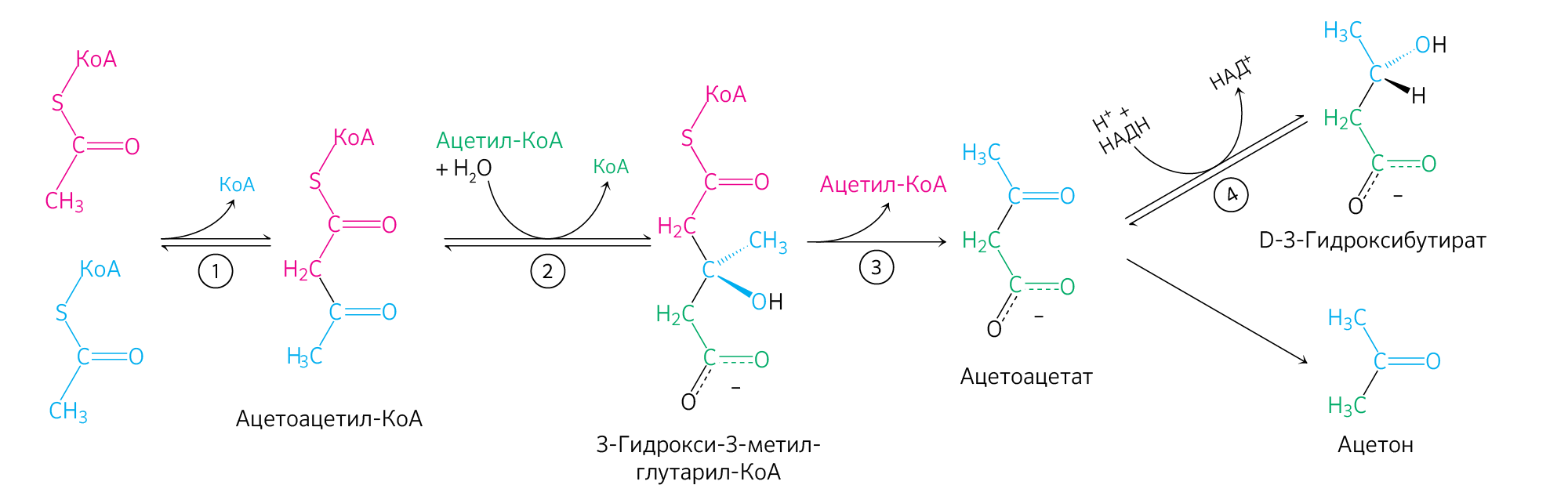
Рисунок 11. Анаболизм кетоновых тел.
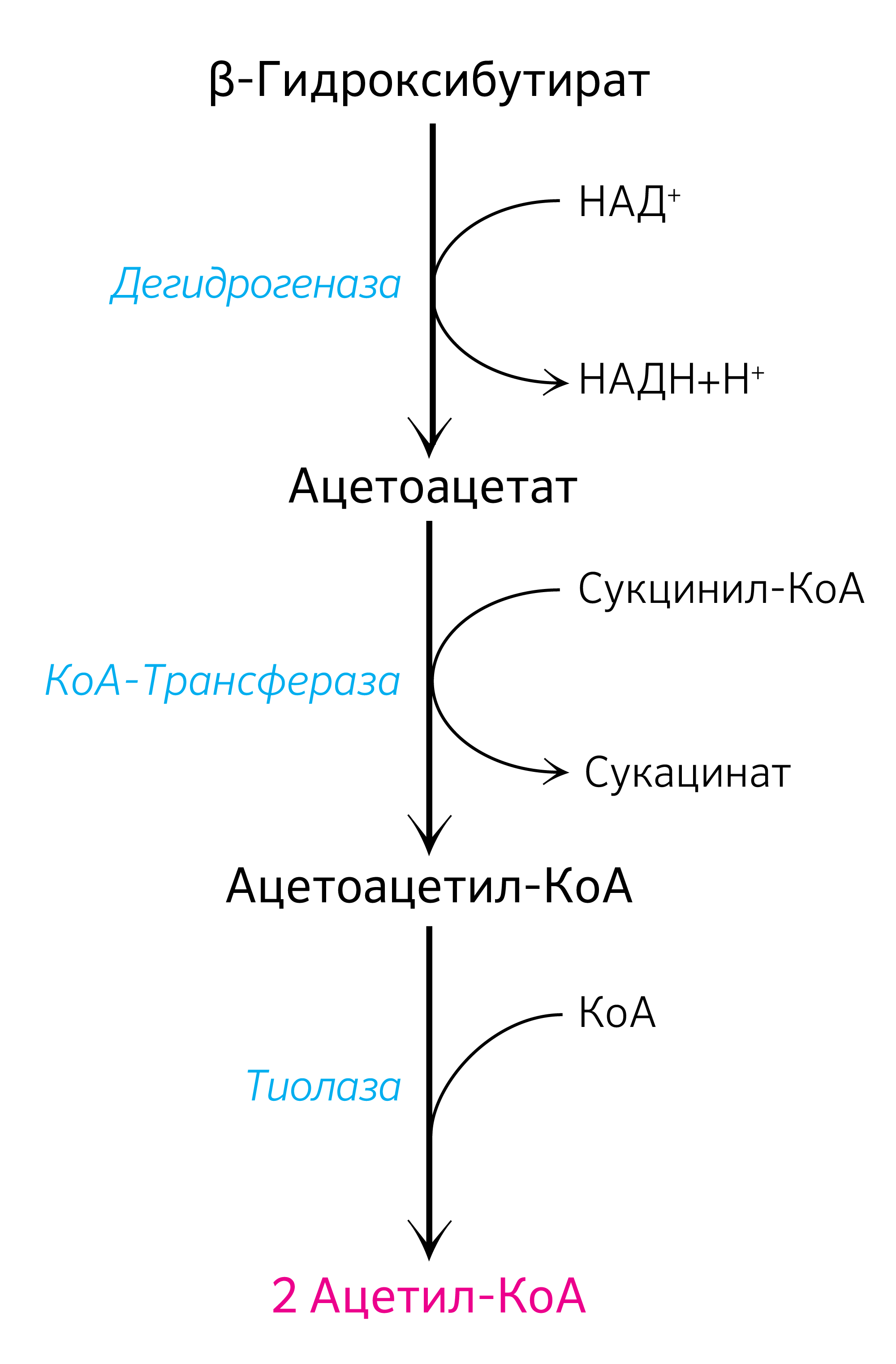
Рисунок 12. Катаболизм кетоновых тел.
Выходит, что белки, жиры и углеводы являются источниками не только ацетил-КоА, который «закрутит колесо ЦТК», но и незаменимых для его стадий субстратов.
Большинство окислительных реакций ЦТК обратимы, но исключение составляют три стадии: превращение оксалоацетата в цитрат, окисление изоцитрата до α-кетоглутарата и окисление α-кетоглутарата до сукцинил-КоА.
Далее подробно остановимся на каждой из восьми стадий:
I стадия — образование цитрата;
II стадия — образование изоцитрата;
III стадия — окисление до α-кетоглутарата;
IV стадия — окисление до сукцинил-КоА;
V стадия — образование сукцината;
VI стадия — окисление до фумарата;
VII стадия — гидратация до малата;
VIII стадия — окисление до оксалоацетата.
СТАДИИ ЦТК
I стадия — образование цитрата
Оксалоацетат (щавелевоуксусная кислота) соединяется с ацетил-КоА в присутствии фермента цитратсинтазы (рисунок 13). Биотин является необходимым компонентом в реакции карбоксилирования пирувата с участием АТФ, в результате чего образуется сам оксалоацетат в присутствии пируваткарбоксилазы, коферментом которой является биотин. Так получается трикарбоновый цитрат — лимонная кислота, поэтому ЦТК иногда называют циклом лимонной кислоты.
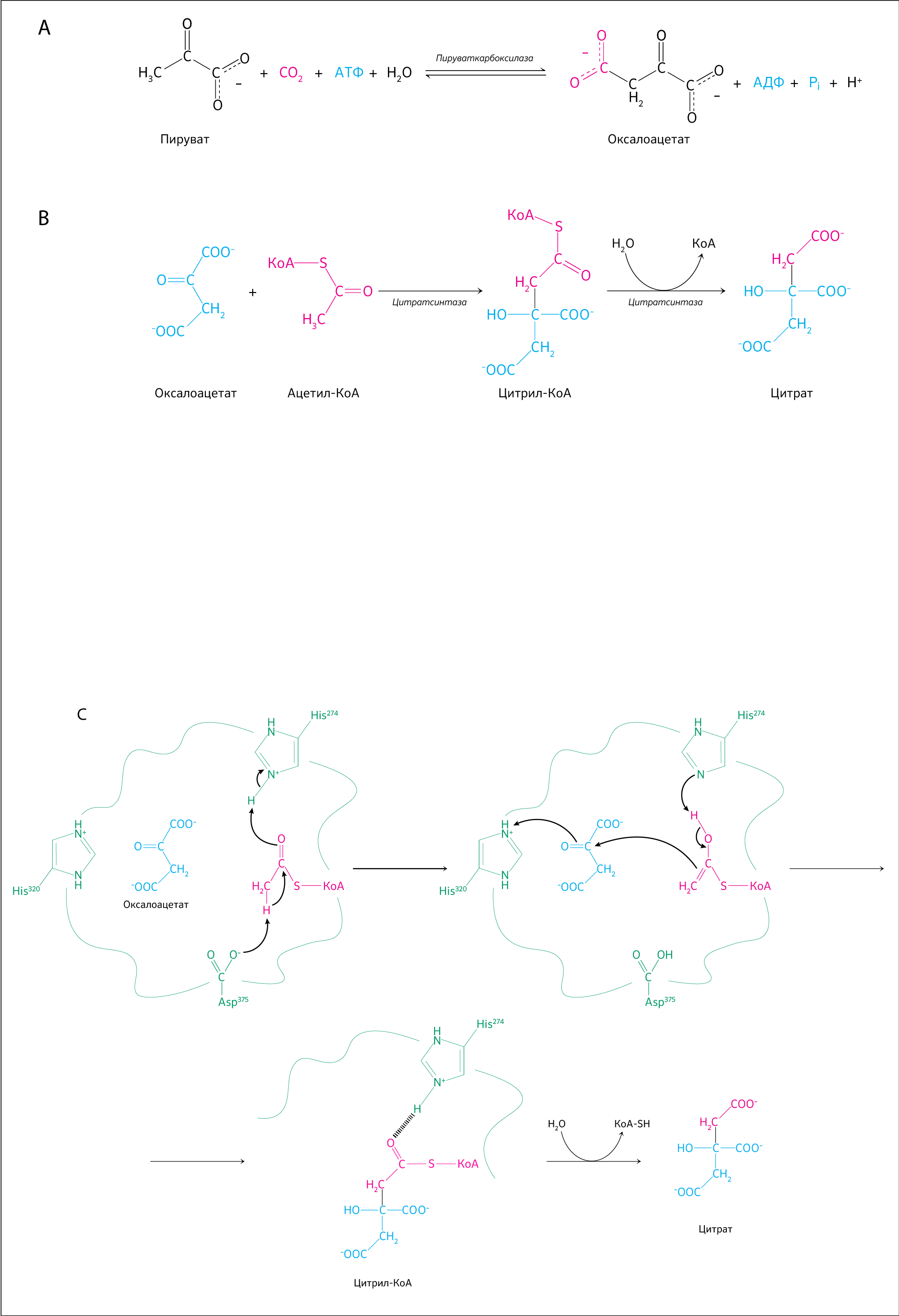
Ацетил-КоА имеет карбонильный (С=О) участок, превращающийся в гидроксильный (С–ОН) благодаря цитратсинтазе (кодируется ядерным геном CIT1). Промежуточный продукт — цитроил-КоА, образующийся в активном центре фермента, — гидролизуется до свободного КоА и цитрата.
Эта стадия ЦТК является одной из немногих необратимых реакций, поэтому уровень экспрессии CIT1 оказывает влияние на остальные стадии цикла.
II стадия — образование изоцитрата через цис-аконитат
Из цитрата необходимо получить его изомер в присутствии фермента аконитазы (аконитатгидратазы), которая будет проводить дегидратацию цитрата и следом гидратацию полученного промежуточного соединения (трикарбоновой кислоты цис-аконитата) для получения изоцитрата, т. е. является одновременно и изомеразой, и гидратазой (рисунок 14).
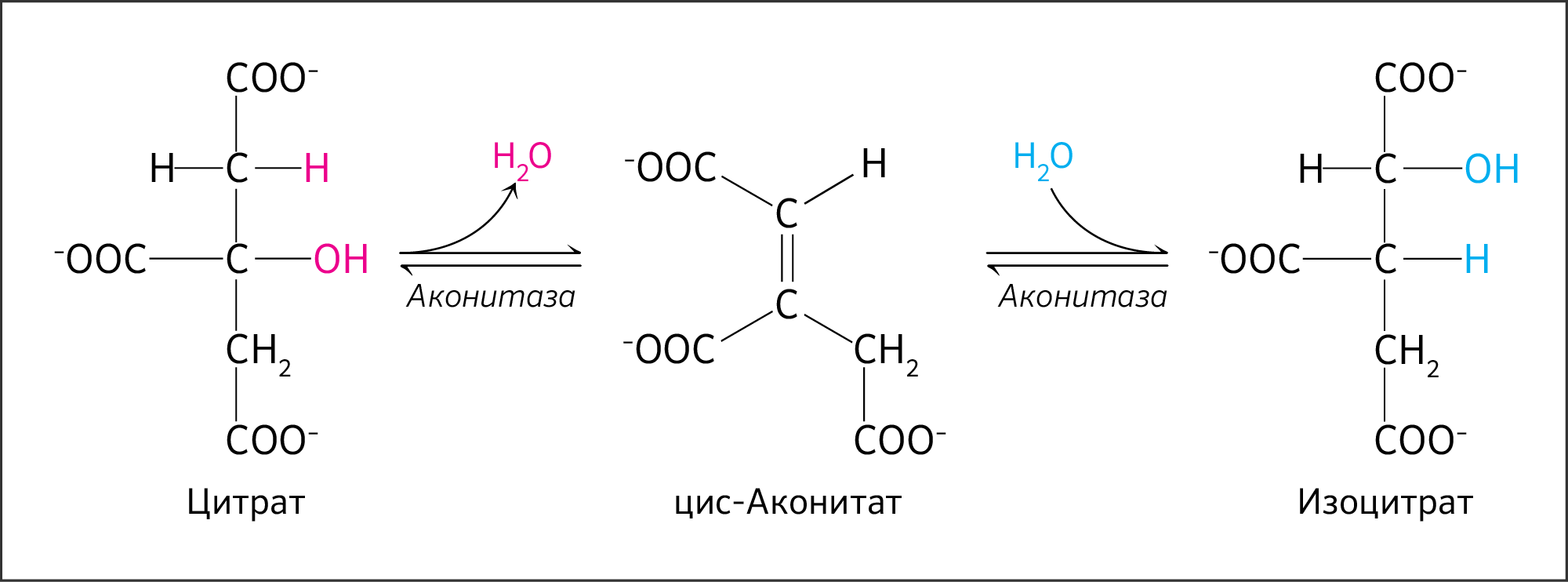
Аконитаза (лат. aconítum — борец, была открыта в растении Aconítum napéllus) — название данного соединения по международной номенклатуре — аконитатгидратаза. Фермент имеет митохондриальную и цитоплазматическую изоформы, кодируемые ядерными генами ACO1 на p-плече 9 хромосомы и ACO2 на q-плече 22 хромосомы соответственно. Митохондриальная изоформа фермента катализирует превращение цитрата в изоцитрат с использованием НАД+ (нарушение гена ACO1 приводит к неспособности провести эту стадию ЦТК [8]), а цитоплазматическая катализирует превращение цитрата в изоцитрат с использованием НАДФ, которая образует НАДФН, участвующий в синтезе ВЖК и стероидов.
III стадия — окисление изоцитрата до α-кетоглутарата
Начиная с этой стадии цикл перестает быть трикарбоновым с точки зрения химических структур последующих субстратов. Здесь фермент изоцитратдегидрогеназа катализирует окислительное декарбоксилирование изоцитрата с образованием α-кетоглутарата (оксоглутарата) и СО2. Реакция проходит через промежуточное соединение — оксалосукцинат — и является необратимой.
Кроме декарбоксилирования в этой реакции происходит восстановление кофермента HAД+ до НАДH+H+ (НАДФН) (рисунок 15).
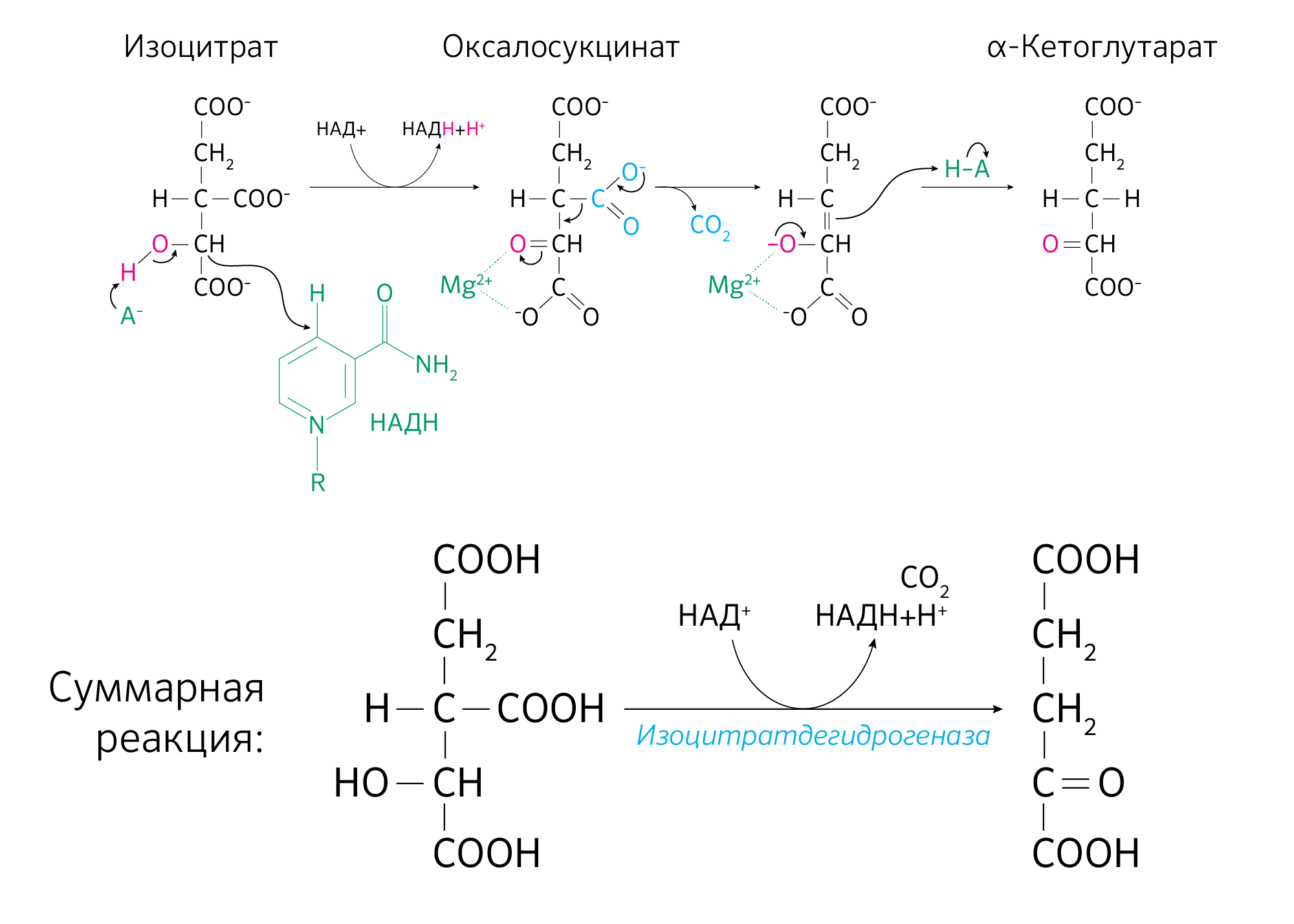
Изоцитратдегидрогеназа является октамером, состоящим из четырех субъединиц Idh1p и четырех субъединиц Idh2p, кодируемых генами второй хромосомы IDH1 (внемитохондриальный) и IDH2 (внутримитохондриальный) соответственно. Гены IDH1 и IDH2 транскрибируются независимо друг от друга, при этом изменение экспрессии любого из них не влияет на транскрипцию другого, но гетерозиготные мутации в любом из генов изоцитратдегидрогеназы приводят к снижению активности фермента с сохранением функции [9].
Кроме того, есть структурно не связанный с IDH1 и IDH2 ядерный ген IDH3, который является интегральным для ЦТК и имеет три изоформы: IDH3A, IDH3B, IDH3G. Суть в том, что IDH1 и IDH2 полагаются на кофермент НАДФ как на акцептор получаемых в ходе окислительного декарбоксилирования электронов, а IDH3 связан именно с НАД+. Поэтому изоцитратдегидрогеназа принадлежит трем отдельным изоформам, одна из которых связана с НАД+, а две другие с НАДФ, но функционально они одинаковы.
Более того, эта стадия является скорость-лимитирующей для всего ЦТК. Окисление изоцитрата до α-кетоглутарата происходит медленно по той простой причине, что IDH3 аллостерически ингибируется образующимся НАДH+H+, α-кетоглутаратом и АТФ, а активируется НАД+, изоцитратом и АДФ.
IV стадия — окисление α-кетоглутарата до сукцинил-КоА
Далее происходит такое же окислительное декарбоксилирование как и с пируватом. Теперь α-кетоглутарат необратимо превращается в сукцинил-КоА и СО2 (рисунок 16). А значит и выполнять окислительное декарбоксилирование будет достаточно похожий фермент — α-кетоглутаратдегидрогеназный комплекс. Он близок к пируватдегидрогеназному комплексу по структуре, функциям и также включает три типа ферментов: КЕ1/КЕ2/КЕ3, каждый из которых кодируется отдельным ядерным геном KGD1, KGD2 и LPD1 соответственно [10].
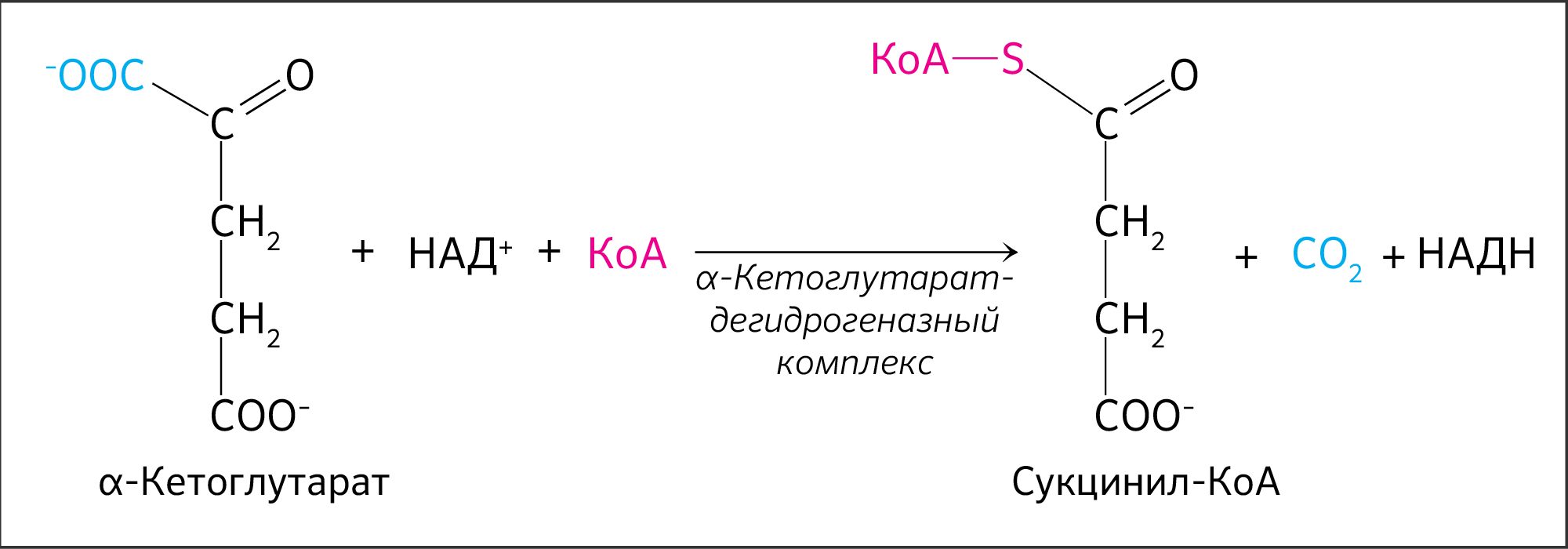
В результате реакции у α-кетоглутарата отщепляется группа –СООН в α-положении. Отщепление кислотного остатка в результате даст CO2 и гидрид-анион. Бывшему α-кетоглутарату к С=О присоединяется свободная теперь группа –SКоА и получается сукцинил-КоА.
V стадия — превращение сукцинил-КоА в сукцинат
Сукцинил-КоА является соединением, которое может формировать новый порядок реакций. Доказательством тому является возможность альтернативного хода ЦТК под названием ГАМК-шунт (рисунок 17) в нейронах и астроцитах центральной нервной системы, где IV стадия заканчивается превращением не в сукцинил-КоА, а в глутамат, который декарбоксилируется в ГАМК (тормозный нейромедиатор). Затем ГАМК метаболизируется сначала до сукцинилового полуальдегида, а потом до сукцината — продукта V стадии.
Образование сукцината происходит при участии фермента сукцинил-КоА-синтетазы (используется лигаза, т. к. катализируется образование новых связей между индивидуальными молекулами с использованием энергии макроэргических соединений) (рисунок 18).
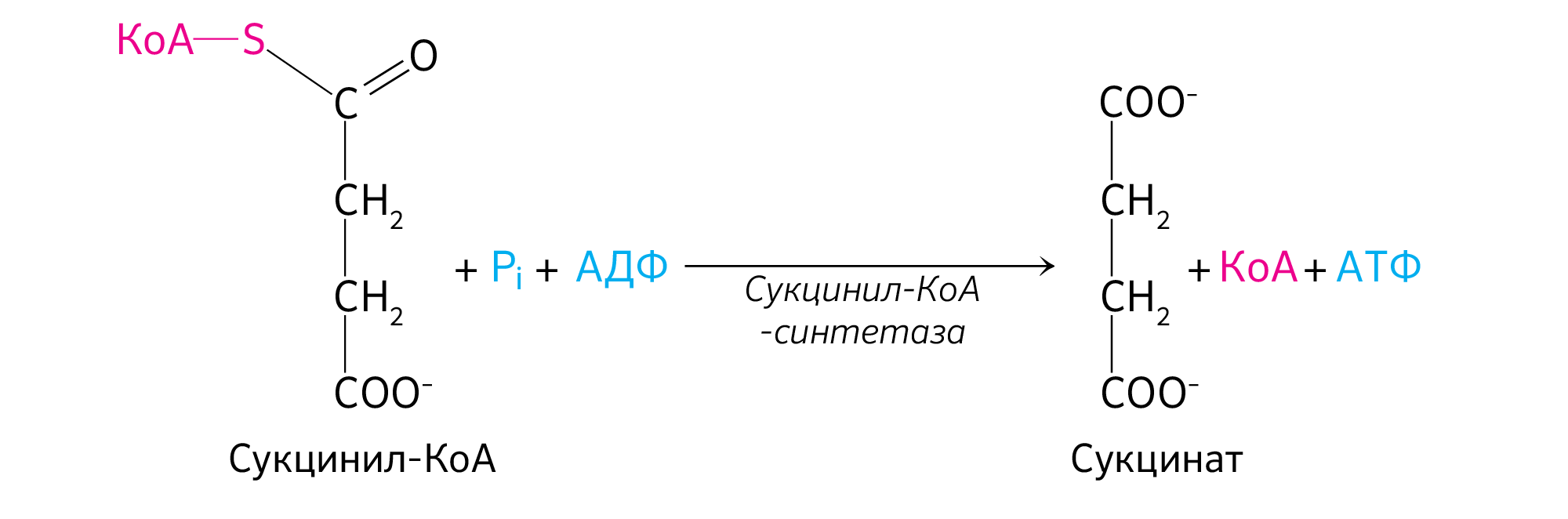
Сукцинил-КоА-синтетаза функционирует в виде гетеродимера и состоит из субъединиц α и β, необходимых для его каталитической активности и кодируемые генами LSC1 и LSC2 соответственно [11]. Он отщепляет –SКоА, H+ для его связки будет взят у H3PO4, а оставшаяся фосфорная группа H2PO4- присоединится к ГДФ или АДФ с образованием ГТФ или АТФ. Образование ГТФ у животных или АТФ у растений за счет энергии, запасенной при окислительном декарбоксилировании α-кетоглутарата до сукцинил-КоА с его тиоэфирной группой, является реакцией субстратного фосфорилирования, как и синтез ATФ при гликолизе (таблица 1).
VI стадия — окисление сукцината до фумарата
Сукцинат (янтарная кислота) окисляется до фумарата под действием оксидоредуктазы — сукцинатдегидрогеназы (у эукариот это митохондриальный хромопротеин и единственный фермент ЦТК, который закреплен во внутренней мембране митохондрий). Активный центр фермента образуют субъединицы, содержащие флавин и железо-серные группы, кодируемые генами SDH1 и SHD2 соответственно [12,13]. Закрепление фермента в мембране митохондрии осуществляется с помощью двух гидрофобных субъединиц, кодируемых генами SDH3 и SDH4 [14,15]. Для сборки функционального комплекса необходим шаперон семейства Hsp60-Tcm62p [16].
В этой реакции от сукцината отщепляется гидрид-анион, но коферментом будет являться не НАД+ или НАДФ, как в предыдущих реакциях, а флавинадениндинуклеотид (ФАД) (образуется из рибофлавина, витамина B2), т. к. этот кофермент является ковалентно связанной простетической группой сукцинатдегидрогеназы. В итоге в исходном соединении просто создается ковалентная связь.
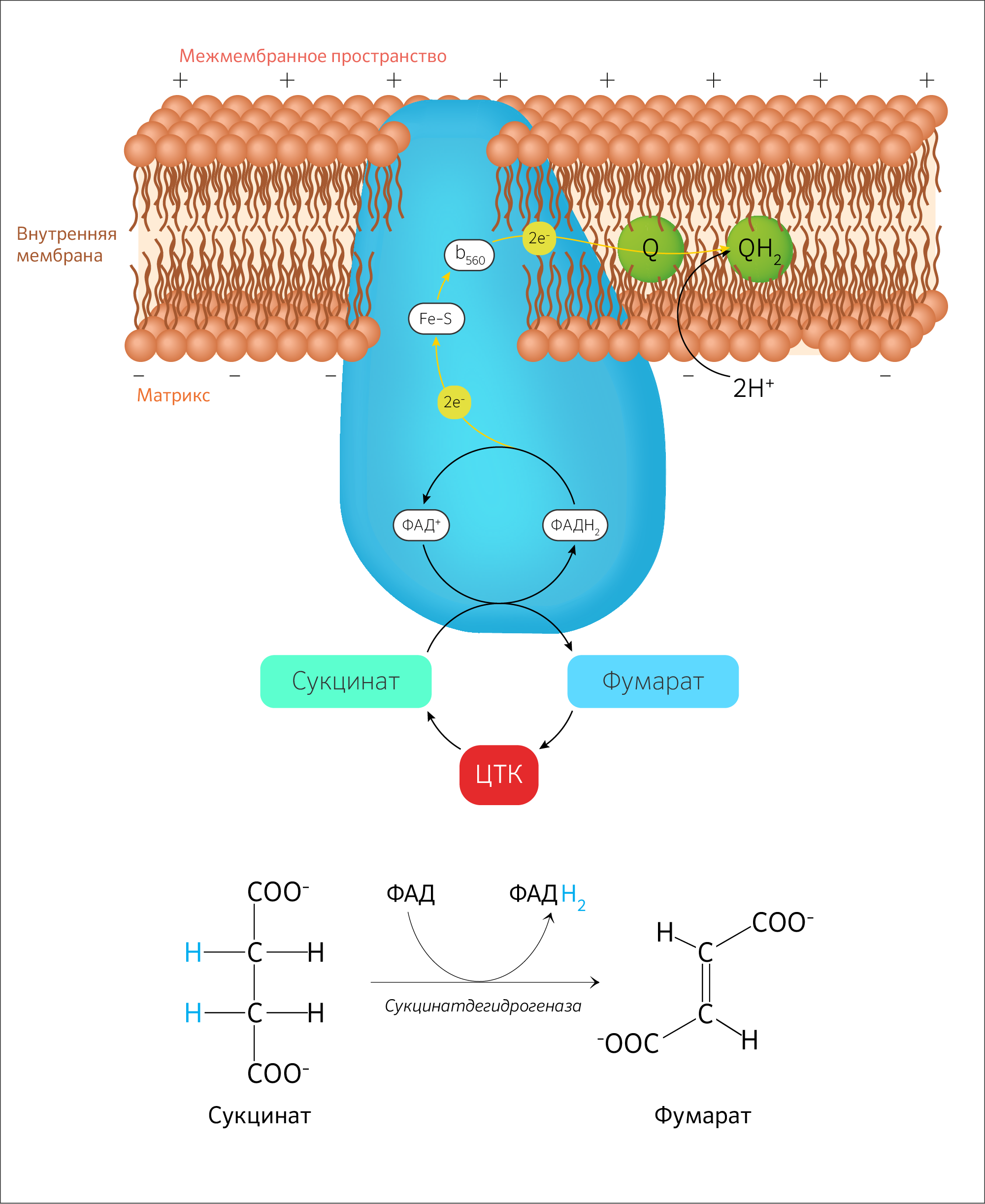
Изучая эту стадию подробно, непременно столкнешься с понятием об убихиноне (кофермент Q) (рисунок 19). Этот компонент ЦПЭ принимает участие в окислительном фосфорилировании как компонент митохондриального ферментного комплекса (МФК). Всего же на сегодня изучено четыре МФК (1 — НАДН-дегидрогеназный комплекс, 2 — сукцинатдегидрогеназа, 3 — убихинон-цитохром С-оксидоредуктаза, 4 — цитохромоксидаза). Функционирование МФК 1 и 3 приводит к генерации активных форм кислорода (АФК).
Таким образом, сукцинатдегидрогеназа является одновременно ферментом этой стадии и обязательным компонентом ЦПЭ. Какова ее функция в ЦПЭ? Дело в том, что НАДH+H+ переносится гидрид-анион, который будет отдаваться МФК 1, а потом МФК 3 и 4, создавая необходимый пул электронов. К слову, вновь о роли аминокислот в организме человека: убихинон синтезируется из аминокислот тирозина, фенилаланина и мевалоновой кислоты (субстратом для синтеза мевалоновой кислоты служит ацетил-КоА).
VII стадия — гидратация фумарата до малата
Полученная ковалентная связь между атомами углерода фумарата будет подвергаться гидратации до малата (L-малата, яблочной кислоты) при действии гомотетрамерного фермента фумаратгидратазы или фумаразы (рисунок 20). Фумараза стереоспецифична к транс-изомерам, а не цис-изомерам.
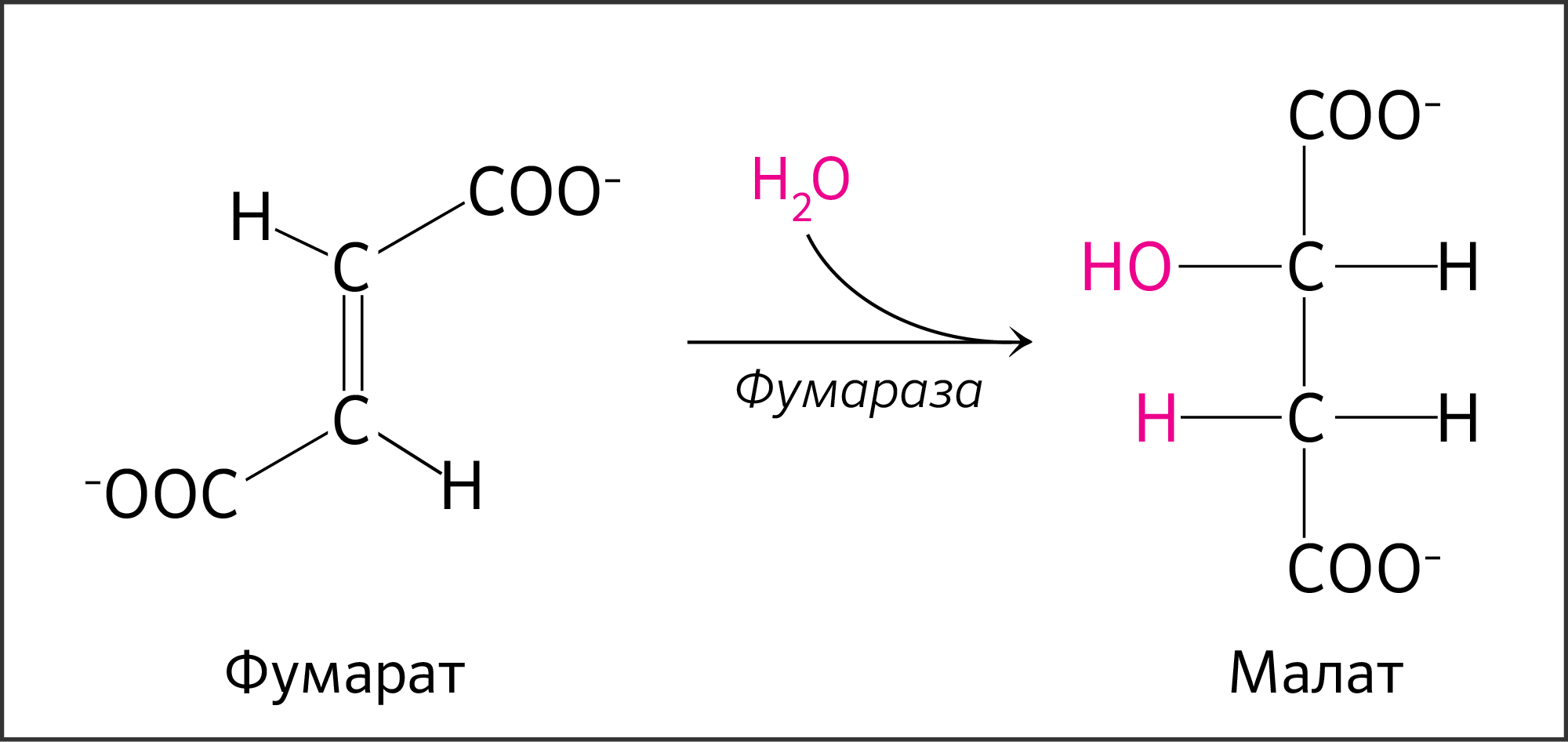
Рисунок 20. Реакция превращения фумарата в малат.
В отличие от других ферментов цикла, имеющих митохондриальную и цитоплазматическую изоформы, которые кодируются отдельными генами, обе изоформы фумаразы закодированы в одном и том же ядерном гене — FUM1 — и транслируются с одного и того же транскрипта [17] путем альтернативного сплайсинга. Фумараза принадлежит ферментам двух классов. Первый класс обнаружен у прокариот, и это термолабильный железозависимый фермент. Второй класс обнаружен у млекопитающих, дрожжей и коринебактерий, где он уже является термостабильным железонезависимым ферментом [18,19].
VIII стадия — окисление малата до оксалоацетата
Последняя стадия ЦТК (рисунок 21).
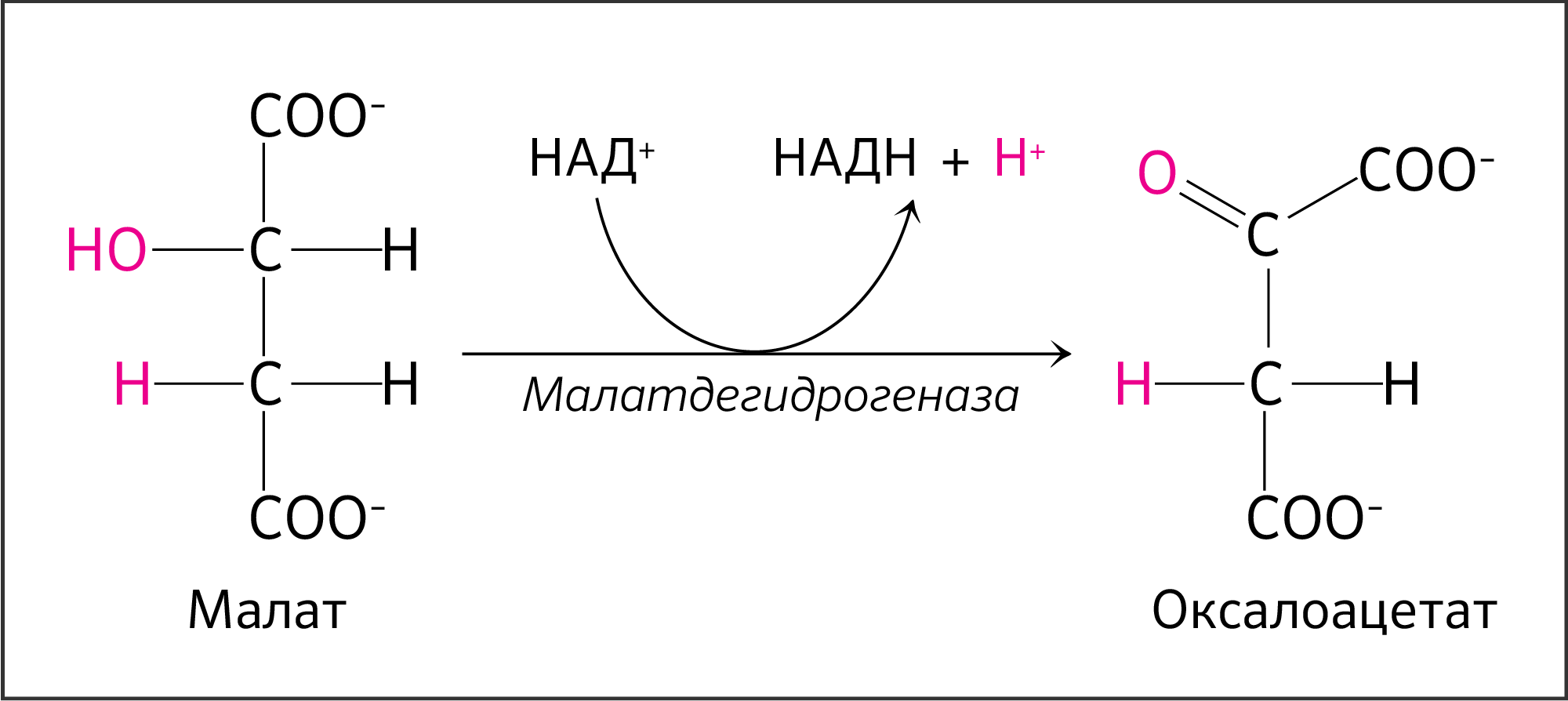
Реакция сопровождается действием уже привычной НАД-зависимой дегидрогеназы — L-малатдегидрогеназы. Митохондриальная малатдегидрогеназа эукариот функционирует в виде гомодимера, субъединицы которого закодированы в митохондриальном гене MDH2 [20].
Концентрация оксалоацетата в митохондриях мала (около 10-6 М). Реакция превращения малата в оксалоацетат при стандартных условиях характеризуется положительной энергией Гиббса (ΔG'° +29,7), что направляет ее в обратном направлении, но благодаря малой концентрации оксалоацетата реакция осуществляется около равновесия (ΔG 0).
ЭНЕРГЕТИЧЕСКАЯ РОЛЬ ЦТК
Освобождающаяся в результате окисления одной молекулы ацетил-КоА энергия в значительной мере сосредоточивается в макроэргических фосфатных связях АТФ. В ходе ЦТК синтезируется также одна молекула ГТФ (субстратное фосфорилирование), что эквивалентно одной молекуле АТФ при действии фермента нуклеозиддифосфаткиназы. Полученные НАДH+H+ понесут гидрид-анионы для работы АТФ-синтазы путем их передачи через 1, 3 и 4 МФК на внутренней мембране митохондрий (кристы).
При этом на 1 НАДH+H+ будет приходится 3 АТФ, а на 1 ФАДН2 придется только 2 АТФ из-за передачи электронов на МФК 1, 3 и 4 ЦПЭ с меньшей энергией, чем у НАД+. Вследствие этого электроны от ФАД-зависимых дегидрогеназ поступают в ЦПЭ на МФК 2, минуя первый пункт сопряжения — НАДН-дегидрогеназный комплекс.
Энергетический выход одного ЦТК на основании энергии Гиббса:
— 3 НАДH+H+ = 9 АТФ
— 1 ФАДН2 = 2 АТФ
— 1 ГТФ = 1 АТФ
Итого: получаем 12 АТФ из одного ацетил-КоА. Так как основным источником энергии являются углеводы, то не стоит забывать, что при гликолизе мы из одной молекулы глюкозы получаем две молекулы пирувата, поэтому смело удваиваем значение АТФ до 24 и прибавляем 2 НАДH+H+ и 2 АТФ, полученных при гликолизе, а также 2 НАДH+H+, полученных в ходе реакций окислительного декарбоксилирования пирувата. В идеально работающей системе при расщеплении в тканях одной молекулы глюкозы получается 38 АТФ согласно уравнению:
С6Н12О6 + 6О2 → 6СО2 + 6Н2О
Однако глицеролфосфатная челночная система переноса НАДH+H+ в матрикс митохондрий из цитоплазмы идёт в конечном счёте с потерей АТФ. Данный механизм будет рассмотрен ниже, но в таком случае при расщеплении одной молекулы глюкозы будет получаться около 25 АТФ.
В энергетическом отношении полное расщепление глюкозы является более эффективным процессом, чем анаэробное дыхание.
Таблица 1. Образование макроэргических фосфатных связей в ходе гликолиза, ЦТК и аэробного дыхания.
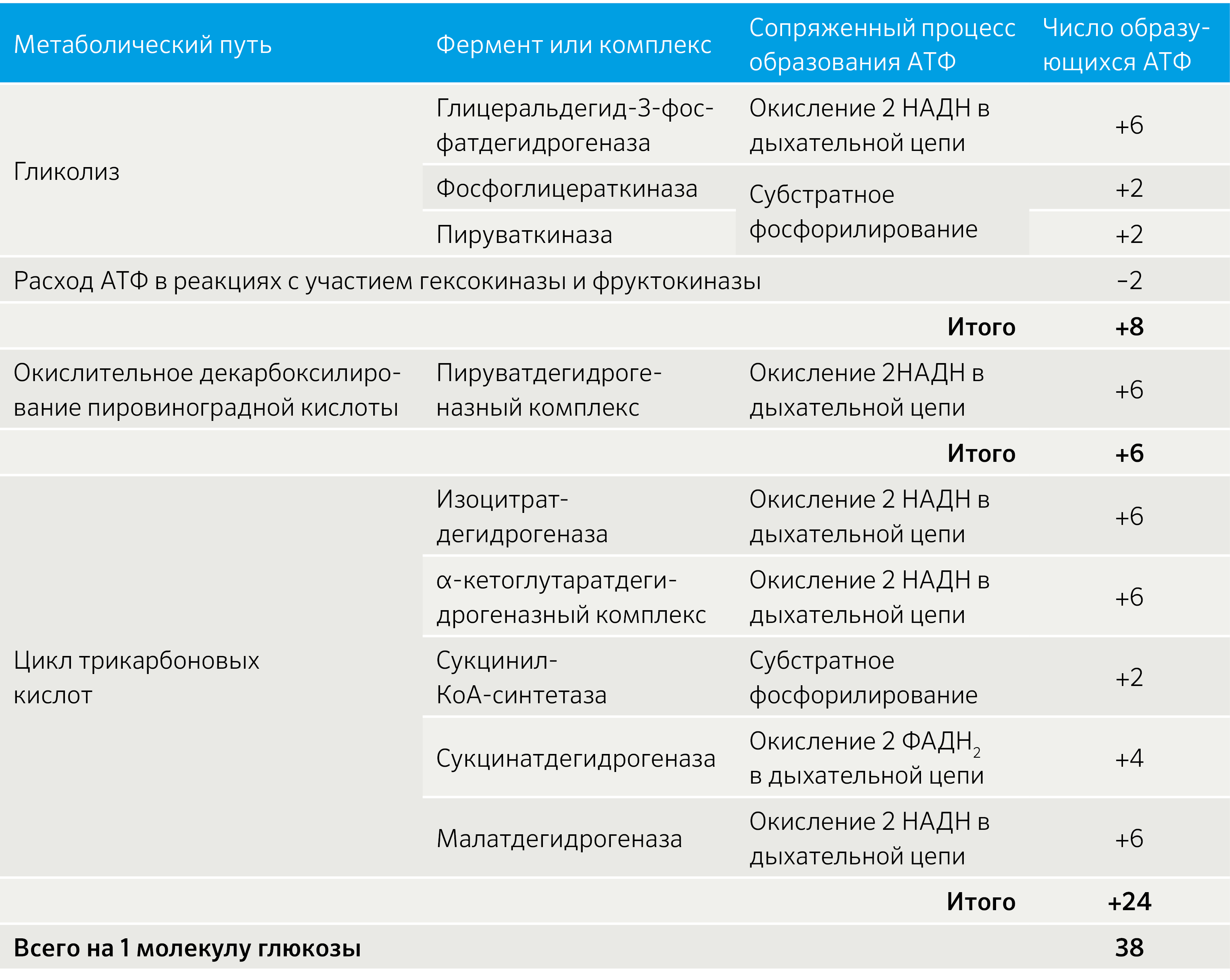
Из вышесказанного следует, что основная роль ЦТК заключается в поставке 4 гидрид-анионов (или 8 электронов — 6 на НАДH и 2 на ФАДH2) в ЦПЭ. Кроме того, в самом цикле образуется одна молекула ГТФ.
РОЛЬ ЦТК В МЕТАБОЛИЗМЕ
ЦТК играет важную роль в процессе анаболизма (рисунок 22): из α-кетоглутарата синтезируется глутамат, а из него глутамин, аргинин, пролин, в свою очередь, из оксалоацетата синтезируется аспартат, который при реакции аминирования образует аспарагин. Сукцинил-КоА как предшественник δ-аминолевулиновой кислоты при его конденсации с глицином будет участвовать в синтезе порфиринов у животных или хлорофилла у растений. Из оксалоацетата в процессе глюконеогенеза будет получена глюкоза.
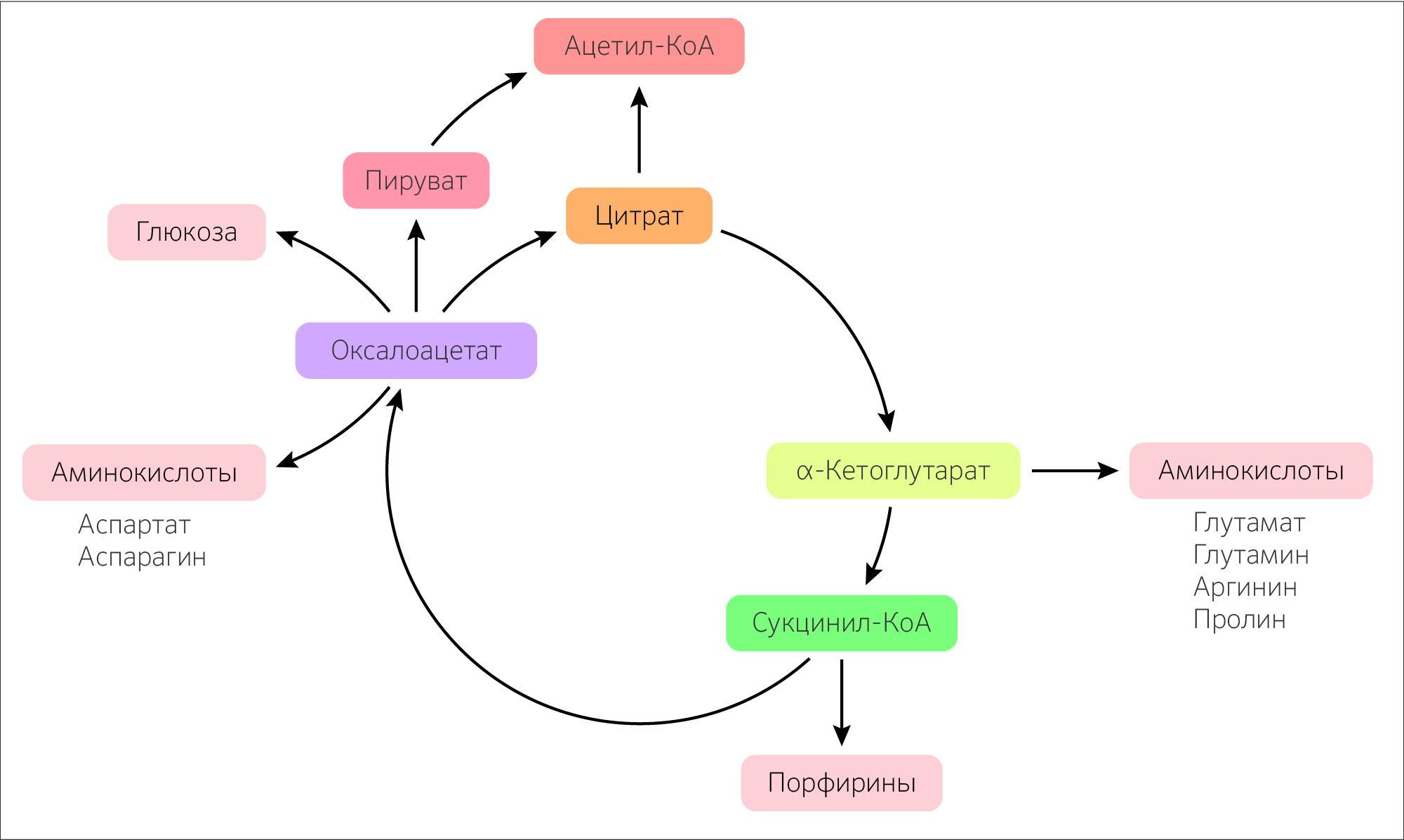
Рисунок 22. Значение цикла трикарбоновых кислот в анаболизме.
В процессе катаболизма (рисунок 23) на I этапе крупные молекулы (полимеры) расщепляются до простых компонентов (мономеров): углеводы превращаются в глюкозу, жиры в ВЖК и глицерол, белки в аминокислоты. На II этапе полученные мономеры внутриклеточно специфически расщепляются до одного и того же метаболита — пирувата. Далее пируват (а также некоторые аминокислоты в процессе дезаминирования и ВЖК в процессе β-окисления) окисляется до ацетил-КоА. III этап представляет собой ЦТК и ЦПЭ (общий путь катаболизма), где образованный ацетил-КоА окончательно распадается до CO2 в митохондриях клетки. То есть 2 атома углерода в составе ацетил-КоА входят в ЦТК (I стадия) и 2 атома углерода в составе CO2 покидают его (III и IV стадии).
Также некоторые аминокислоты могут превращаться в субстраты стадий ЦТК: аргинин, гистидин и глутамат в α-кетоглутарат, а фенилаланин и тирозин в фумарат.
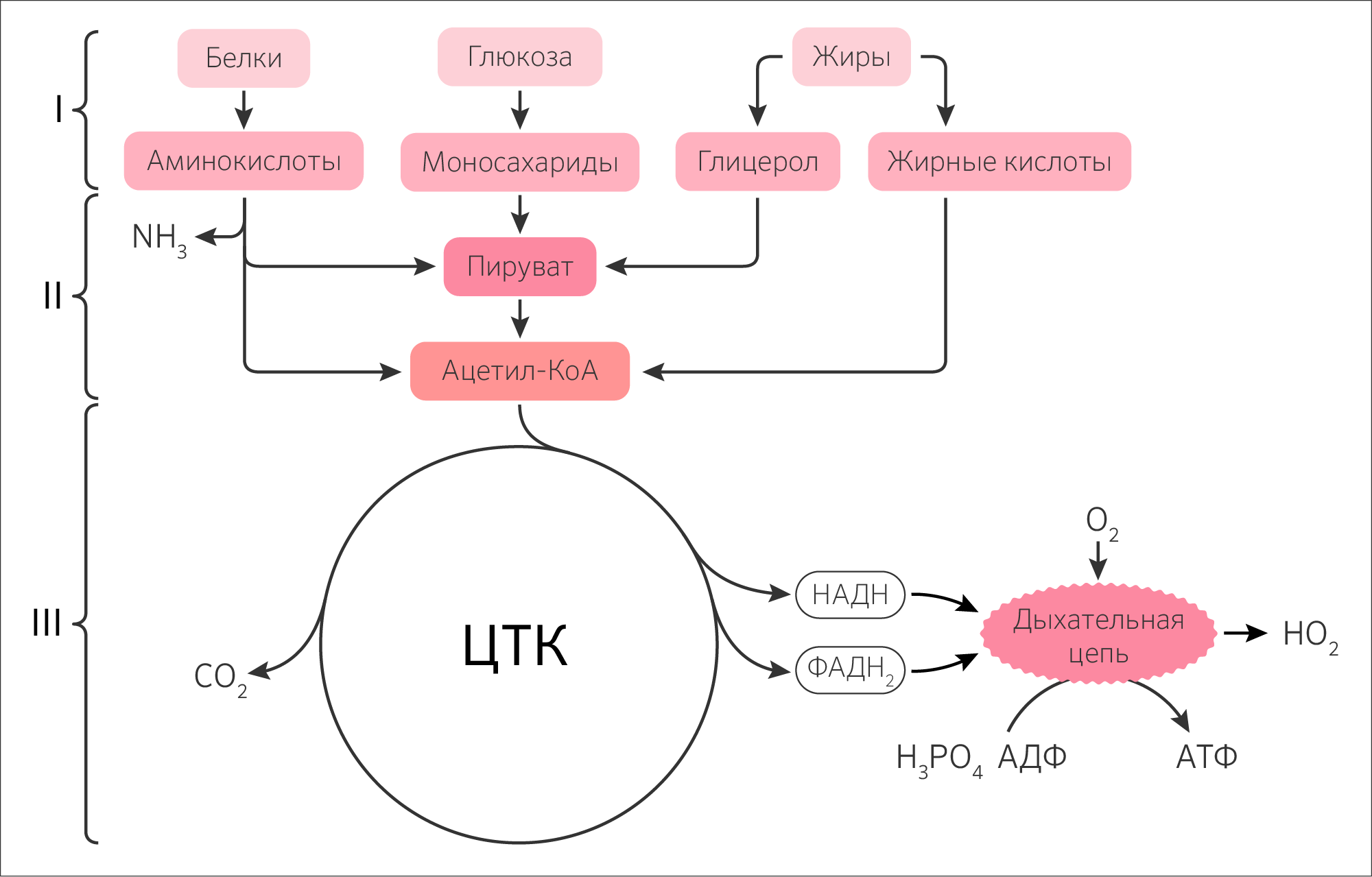
Рисунок 23. Значение цикла трикарбоновых кислот в катаболизме.
Существует связь между ЦТК и циклом мочевины. Такое пересечение названо «бициклом Кребса» (рисунок 24). Непосредственный путь, связывающий ЦТК и цикл мочевины, называется аспартат-аргининосукцинатный шунт: в нем утилизируются аминогруппы. Фумарат, образующийся в аргининосукцинатной реакции в межмембранном пространстве, является субстратом ЦТК. При этом фумарат либо в цикле мочевины, либо при синтезе пуринов может быть превращен в малат, который будет транспортирован в матрикс через малат-аспартатный переносчик для участия в ЦТК (рисунок 25).
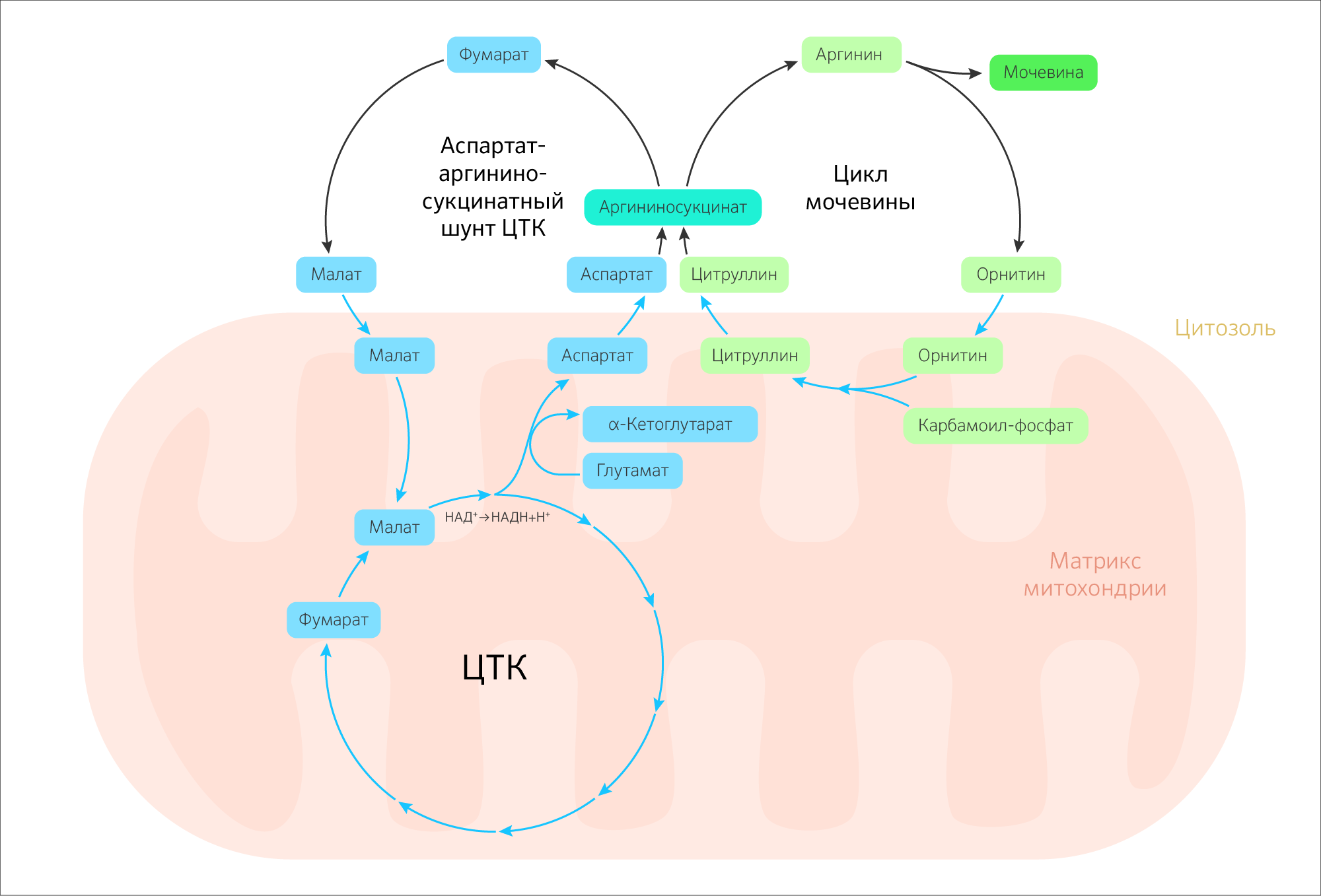
Рисунок 24. Схема взаимодействия цикла трикарбоновых кислот и цикла мочевины (цикл Кребса-Гензелейта).
Ключевым путём транспорта НАДH+H+ из цитоплазмы через внутреннюю мембрану митохондрии в матрикс являются митохондриальные челноки: малат-аспартатная и глицеролфосфатная челночные системы.
Малат-аспартатная челночная система осуществляет перенос НАДH+H+ из межмембранного пространства в митохондриальный матрикс и действует в митохондриях печени, почек и сердца. НАДH+H+ передает гидрид-анион на оксалоацетат, и образуется малат, который переносится через внутреннюю мембрану митохондрии малат-α-кетоглутаратной транспортной системой. В ЦТК малат превращается в оксалоацетат при действии митохондриальной малатдегидрогеназы. Оксалоацетат сам по себе не может вернуться обратно в межмембранное пространство, но может подвергаться действию трансаминазы и превращаться в аспартат, который переносится в межмембранное пространство глутамат-аспартатной транспортной системой. Там аспартат снова переходит в оксалоацетат, который вновь запускает челночную систему.
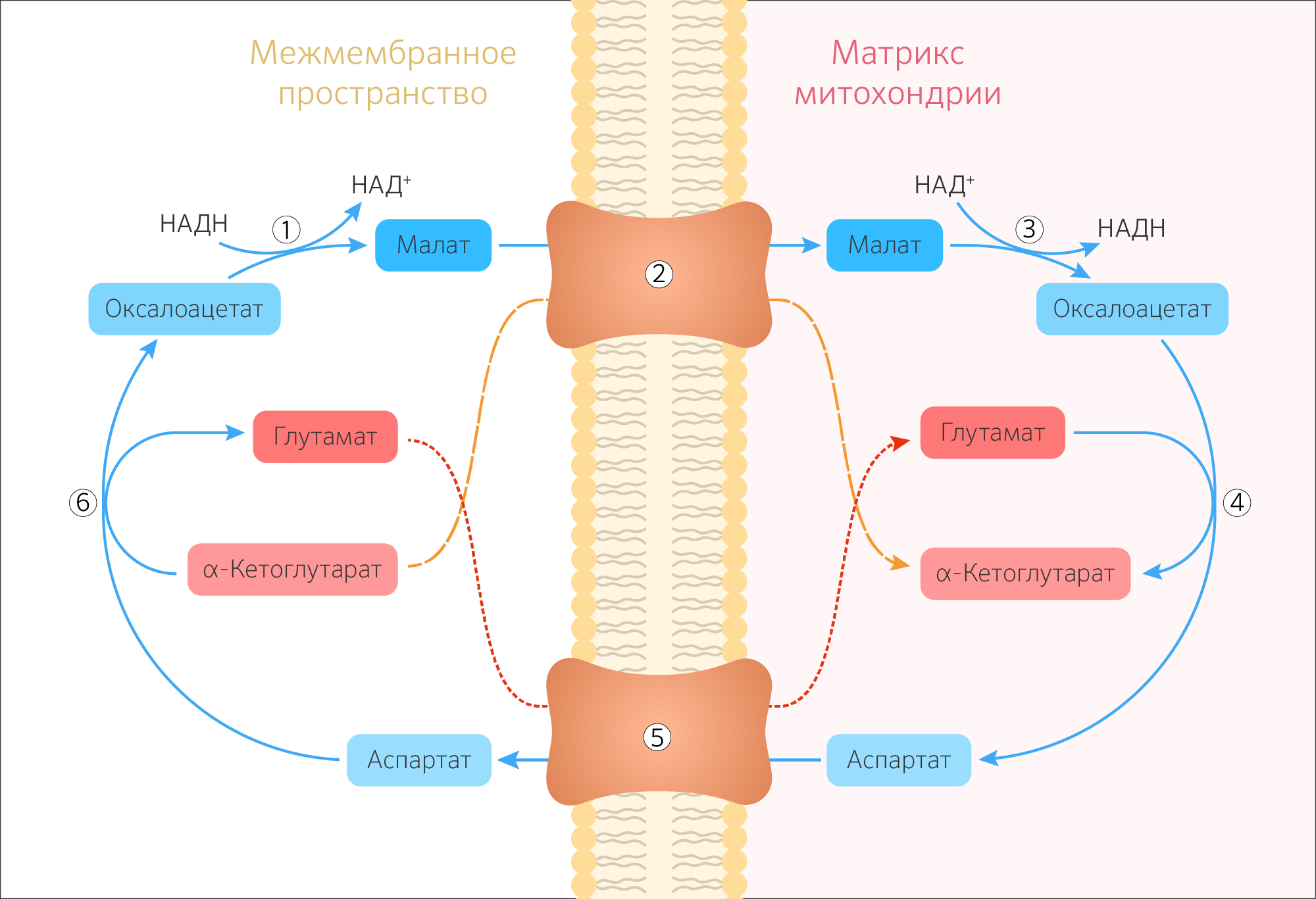
Рисунок 25. Схема малат-аспартатной челночной системы
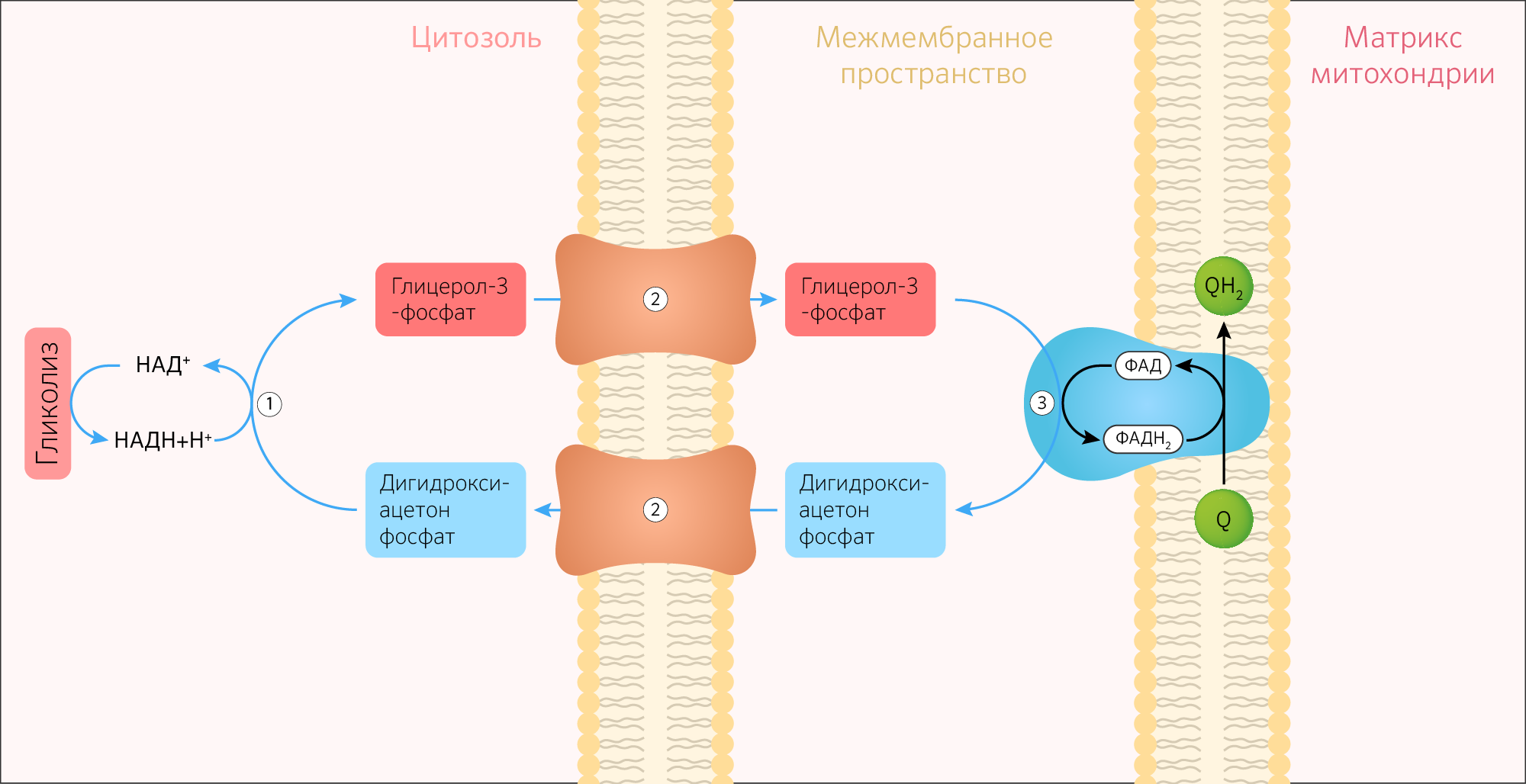
Помимо малат-аспартатной челночной системы в митохондриях работает глицеролфосфатная челночная система (рисунок 26), которая переносит НАДH+H+ из цитоплазмы внутрь митохондрии благодаря ферменту глицерол-3-фосфат-дегидрогеназа. Глицерол-3-фосфат-дегидрогеназа имеет две изоформы — цитоплазматическую и митохондриальную, кодируемые генами GPD1 и GPD2 соответственно. Ключевая разница между двумя изоформами в использовании разных коферментов: у цитоплазматической — НАД+, а у митохондриальной — ФАД.
Цитоплазматическая глицерол-3-фосфат-дегидрогеназа полностью находится в цитоплазме и не проникает в мембрану митохондрии. Митохондриальная глицерол-3-фосфат-дегидрогеназа располагается на внешней стороне внутренней мембраны митохондрии и именно она окисляет глицерол-3-фосфат до ФАДH2, а образующиеся в ходе такой окислительно-восстановительной реакции H+ далее переходят на убихинон ЦПЭ. В результате работы глицеролфосфатной челночной системы на 1 НАДH+H+ будет приходиться только 1,5 АТФ.
Следовательно, ЦТК — это амфиболический цикл. С одной стороны, присутствуют выраженные катаболические процессы, но вместе с тем промежуточные продукты ЦТК начинают новые биосинтетические пути, которые приводят к снижению их концентрации. Такое истощение пула промежуточных продуктов должно пополняться путем анаплеротических реакций.
РЕГУЛЯЦИЯ ЦТК
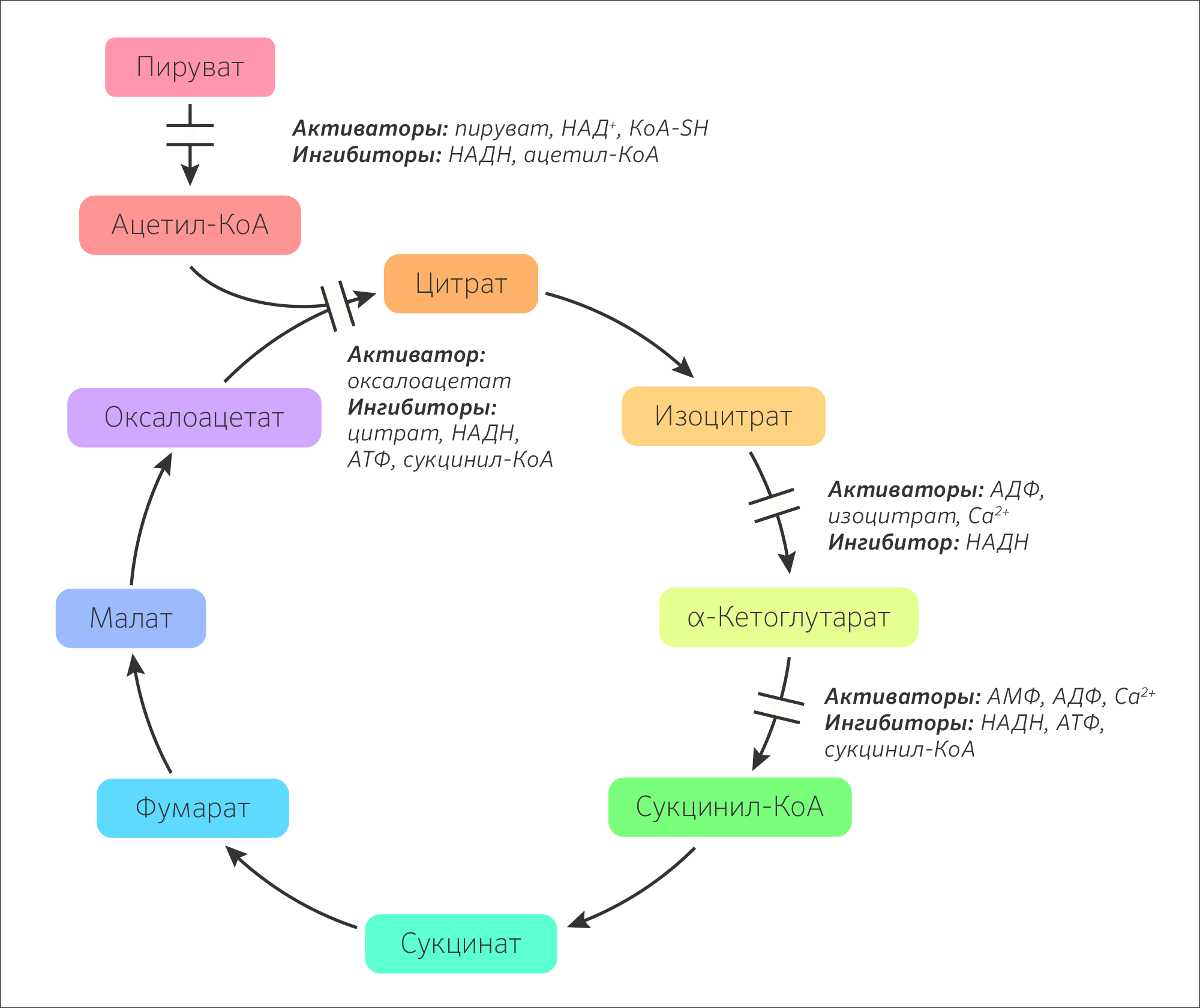
Рисунок 27. Естественная регуляция активности ферментов цикла трикарбоновых кислот по принципу обратной связи.
Скорость реакций ЦТК зависит от обеспеченности клеток энергией АТФ. Активность зависит от соотношения [АТФ]/[АДФ] и [НАДH+H+]/[НАД+] (рисунок 27). То есть общий путь катаболизма активируется при низком энергетическом потенциале клетки или ингибируется при высоком. Соотношение [АТФ]/[АДФ] характеризует энергетический заряд клетки (ЭЗК) по Аткинсу, который выражается формулой:
([АТФ]+1\2[АДФ])/([АТФ]+[АДФ]+[АМФ])
ЭЗК может меняться от 0 до 1. Метаболические пути, приводящие к синтезу АТФ, ингибируются высоким значением ЭЗК, а ведущие к затрате АТФ, активируются высоким значением ЭЗК.
Для ферментов необратимых реакций: пируватдегидрогеназного комплекса, цитратсинтазы, изоцитратдегидрогеназы и α-кетоглутаратдегидрогеназного комплекса индукторами являются такие субстраты как АДФ, окисленный НАД+, а ингибиторами продукты реакции — АТФ, восстановленный НАД+, сукцинил-КоА.
В качестве подтверждения такой зависимости от коферментов и макроэргов рассмотрим некоторые аспекты физиологии и фармакологии. Разобщители тканевого дыхания и окислительного фосфорилирования, например, тиреоидные гормоны, катехоламины, белок термогенин и некоторые антибиотики или лекарственные препараты — парацетамол, динитрофенол, дикумарин — однозначно противостоят депонированию энергии в АТФ. То есть энергия переноса Н+ просто рассеивается в виде тепла из-за того, что при действии разобщителей протоны минуют каналы FОF1-ATФсинтазы (рисунок 28) [21]. Но окислительное фосфорилирование не может происходить, если при этом нет фосфорилирования АДФ с образованием АТФ. Для этого в каналы FОF1-ATФсинтазы должны поступать Н+, так как источником энергии для работы FОF1-ATФсинтазы является трансмембранная разность электрохимических потенциалов. В результате разобщения возрастает концентрация АДФ, потребление O2, окисление НАДН+Н+ и ФАДН2, что приводит к высвобождению тепловой энергии.
Трансмембранный белок термогенин синтезируется в клетках бурой жировой ткани, но гомологичные белки присутствуют и в других тканях. При охлаждении организма эти клетки получают сигнал от симпатической нервной системы, и в них активируется процесс липолиза, что приводит к получению НАДН+Н+ и ФАДН2. Далее активируется ЦПЭ и возрастает электрохимический градиент. Но в мембранах митохондрий клеток бурого жира много темогенина, поэтому большая часть энергии Н+ рассеивается в виде тепла, что и помогает поддерживать температуру тела при охлаждении.
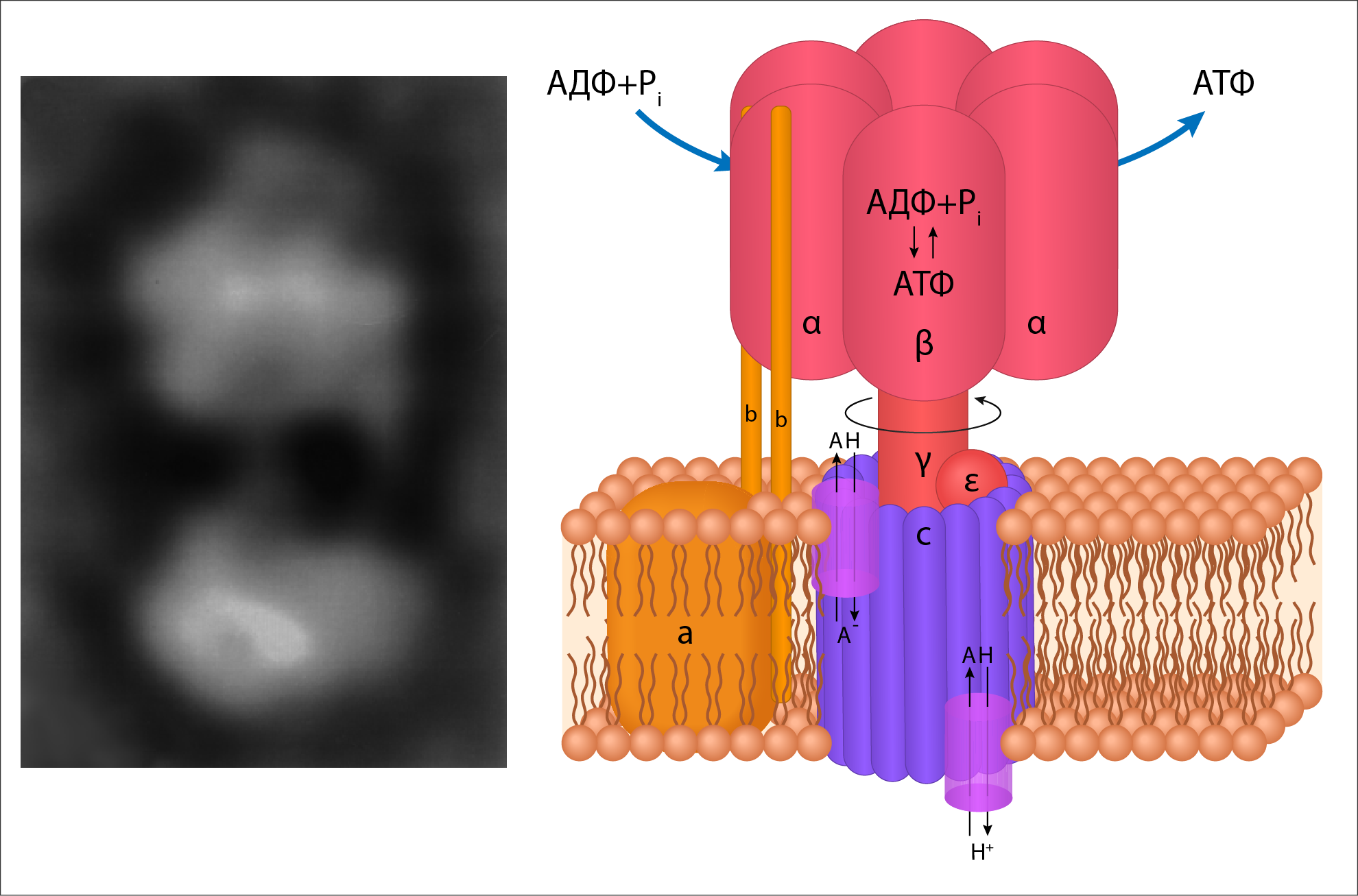
Рисунок 28. Схематическое изображение F1FО-АТФсинтазы и ее электронный снимок. F1 — внемембранный (матриксный) комплекс АТФсинтазы. FО — трансмембранный домен.
Динитрофенол — вещество для производства антисептиков, красителей, гербицидов — проникает в межмембранное пространство митохондрий, где концентрация Н+ выше, и далее с помощью диффузии попадает в матрикс, где концентрация Н+ ниже. Там динитрофенол теряет свой Н+, переходит в ионизированную форму и снова возвращается в межмембранное пространство. Тем самым он рассеивает протонный градиент на внутренней мембране митохондрии. Схожим образом динитрофенол может нарушать глиоксилатный цикл — анаплеротический путь ЦТК у растений, грибов, бактерий и простейших.
Особенно выражено действие парацетамола, а точнее метаболита его действующего вещества — ацетаминофена, на митохондрии гепатоцитов [22]. При поражении свободными радикалами этого вещества митохондриальных мембран, образуемые продукты липопероксидации и свободные ионы кальция нарушают трансмембранный потенциал. Далее при нарушении целостности митохондрий происходит провоспалительный сигналинг цитокинами — IL-1β, ФНО-α, а также простагландина Е2 и тромбоксана В2, что в конечном итоге приводит к деструкции ДНК [23].
Особое место в регуляции ЦТК занимает ответная регуляция пируватдегидрогеназного комплекса. Его активность как фермента зависит от доступности пирувата, аллостерического эффекта и ковалентной модификации. Ковалентная модификация пируватдегидрогеназного комплекса осуществляется путем фосфорилирования и дефосфорилирования по остаткам серина при помощи вспомогательных белков протеинкиназы и фосфопротеинфосфатазы. Протеинкиназа фосфорилирует пируватдегидрогеназный комплекс и инактивирует его, а фосфопротеинфосфатаза дефосфорилирует, превращая его в активную форму. Активность вспомогательных белков может изменяться аллостерически (рисунок 29).
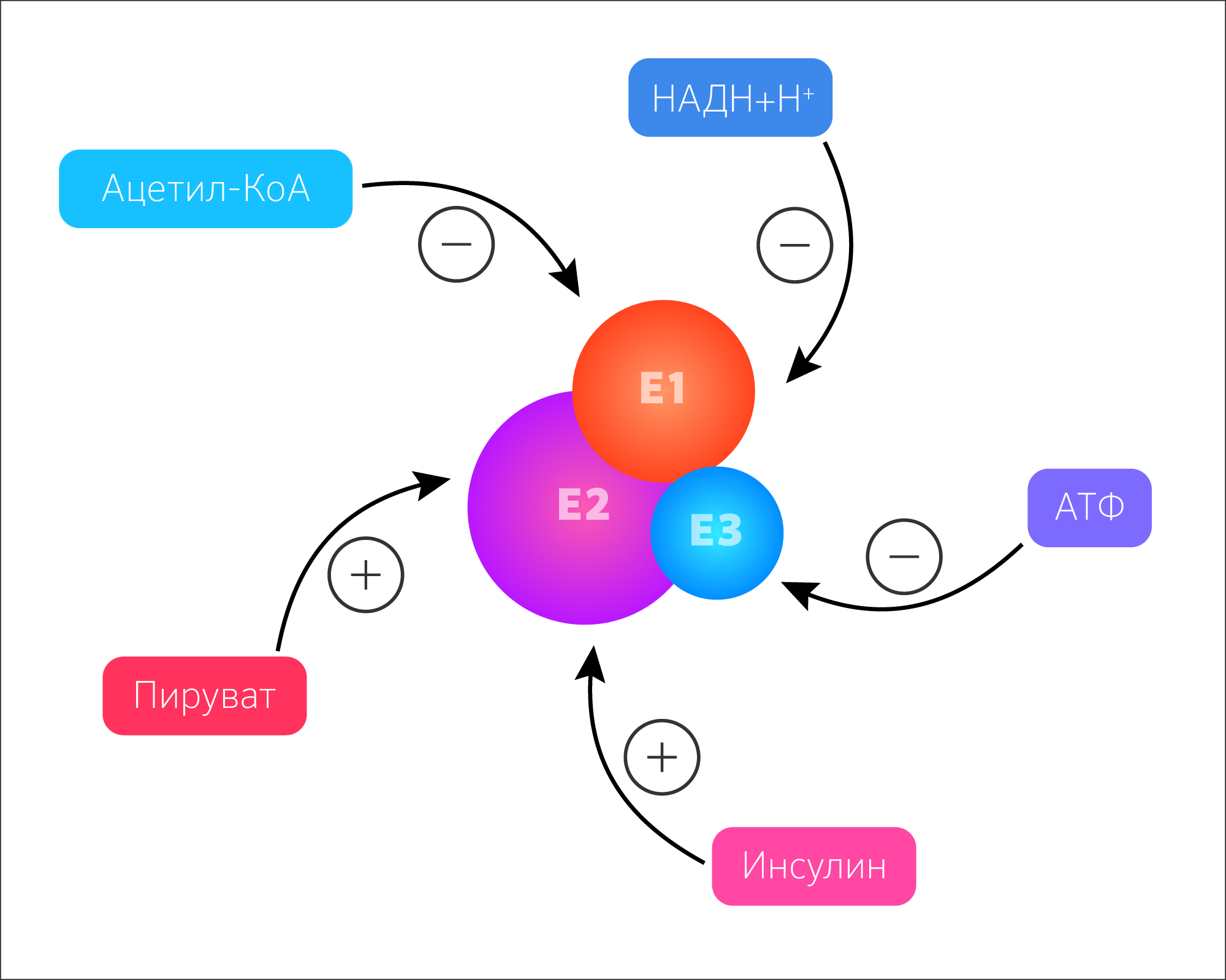
Фосфопротеинфосфатаза активируется при повышении концентрации АДФ, внутриклеточного Ca2+ и под влиянием инсулина за счет повышения концентрации внутримитохондриального Ca2+ . Этот механизм активации пируватдегидрогеназного комплекса играет важную роль в мышцах и жировой ткани.
Таким образом, активность пируватдегидрогеназного комплекса подавляется нарастающей концентрацией АТФ ([АТФ]/[АДФ]), ацетил-КоА (или жирных кислот для осуществления альтернативных путей образования ацетил-КоА) и НАДH+H+ ([НАДH+H+]/[НАД+]). Аллостерическое подавление активности пируватдегидрогеназного комплекса усиливается в присутствии жирных кислот, АМФ, КоА и НАД+, когда в цикл трикарбоновых кислот поступает меньше ацетата.
Помимо вышесказанного нельзя не упомянуть значение многочисленных сигнальных белков в регуляции МФК (рисунок 30). Примечательна роль комплексов ТОМ (транслоказа внешней мембраны) на внешней мембране и TIM (транслоказа внутренней мембраны) на внутренней в переносе белков из цитоплазмы в митохондрию, т. к. уже было сказано, что некоторые изоформы ферментов ЦТК синтезируются ядерной, а не митохондриальной ДНК, и заведомо должны быть не только транспортированы между двумя органеллами, но и специфически внедрены через мембрану митохондрии.
Митохондриальные белки после трансляции переносятся к внешней мембране, где их N-концевая сигнальная последовательность сначала взаимодействует с компонентом ТОМ-комплекса и укрепляется белком TOM-22, который содержит кислые цитозольные домены, взаимодействующие с остатками основных аминокислот в составе N-концевой сигнальной последовательности. После переноса через канал в межмембранном пространстве эта основная часть белка-субстрата за счет сил электростатического притяжения входит в контакт с кислыми доменами белков TOM-22 и TIM-23, но близость друг к другу TOM-22 и TIM-23 обеспечивает прохождение белка-субстрата между этими комплексами, минуя выход в межмембранное пространство.
Непосредственное участие в митохондриальном сигналинге принимает AAA (ATPases associated with various cellular activities — АТФ-синтазы, связанные с различными клеточными активностями) — домен, состоящий из α и β субъединиц мембранно-связанного или FO комплекса АТФ-синтазы, который несет так называемый «Walker motifs» [24] — высоко консервативные трехмерные структуры белка, которые регулируют функциональную деятельность FOF1-АТФ-синтазы. При таргетинге митохондриальные белки находятся в несвернутом состоянии и подготавливаются к транслокации за счет связи с белком HSP70, находящемся в цитозоле. Белки семейства HSP экспрессируются при повышении температуры и помогают транспорту других белков, стабилизируя их в частично свернутом состоянии.
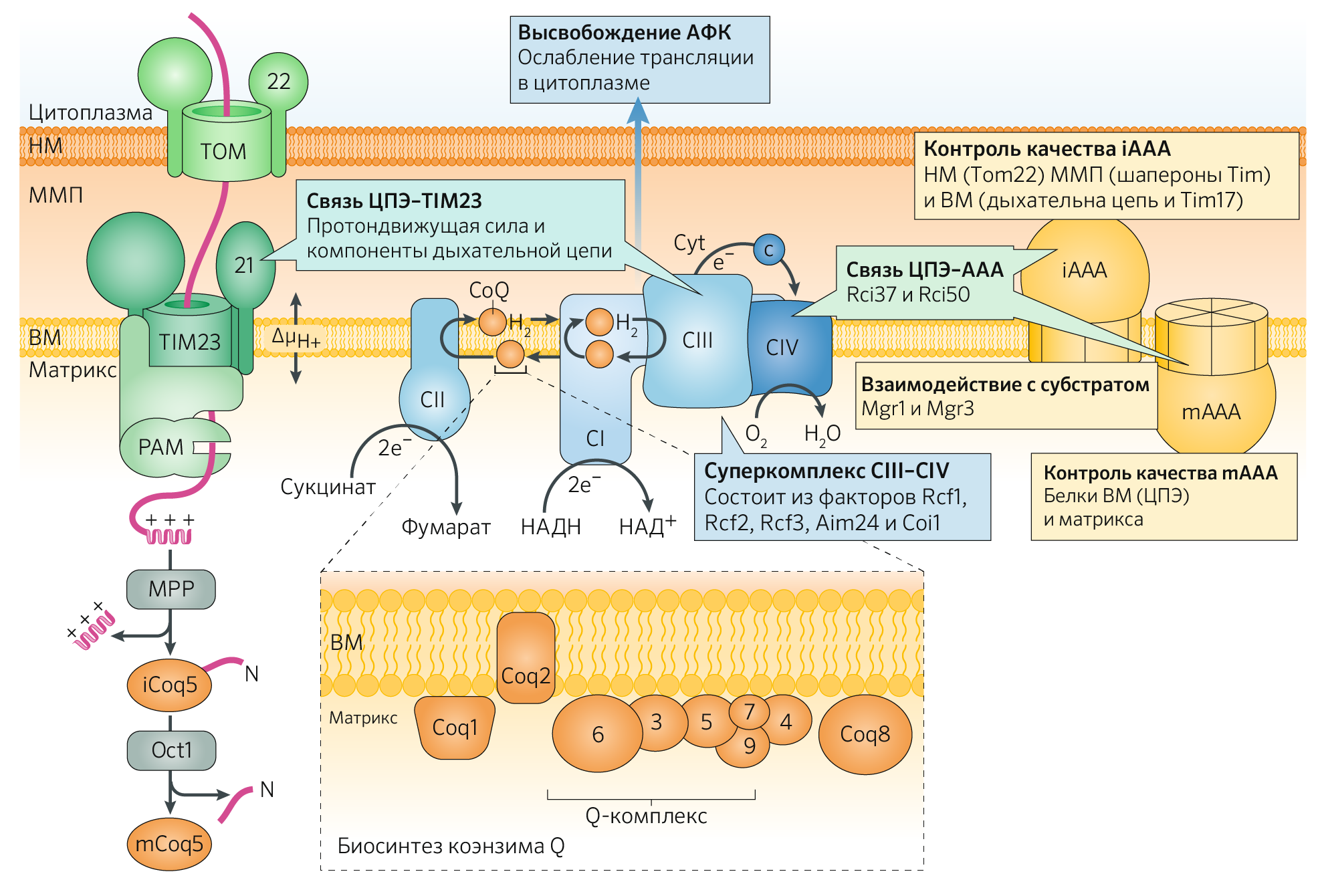
Иными словами, ЦТК и ЦПЭ позволяют митохондрии выполнять ее энергетическую роль — синтез АТФ. Но накопилось много данных, которые говорят об участии митохондрий в регуляции различных сигнальных путей в клетке. К тому же, митохондрии являются одним из основных источников внутриклеточных АФК, и в них содержится большое количество ферментов, катализирующих окислительно-восстановительные реакции. Субстраты этих ферментов окисляются кислородом и превращаются в супероксидные радикалы — предшественники прочих АФК, которые в свою очередь участвуют в регуляции как путем прямого окисления функциональных биомолекул, так и путем активации сигнальных каскадов, например, антиоксидантных систем клетки [26,27].
Основные пути регуляции экспрессии генов, кодирующих ферменты ЦТК
Экспрессия генов (таблица 2), кодирующих ферменты (рисунок 31), зависит от функционирования следующих регуляторных систем:
- глюкозная катаболическая репрессия;
- митохондриальные факторы.
У клеток с дисфункциями митохондрий экспрессия генов ЦТК зависит от белков семейства Rtg. Белок Rtg 3, содержащий bHLH-домен, образует комплекс с белком Rtg 1 в ядре. Этот комплекс участвует в ретроградном ответе клетки, то есть в изменении экспрессии ядерных генов в ответ на нарушение состояния митохондрий, в частности ЦПЭ [28]. Было показано, что Rtg-путь активирует экспрессию генов ферментов первых трех стадий ЦТК [29].
Таблица 2. Регуляция экспрессии генов цикла трикарбоновых кислот.
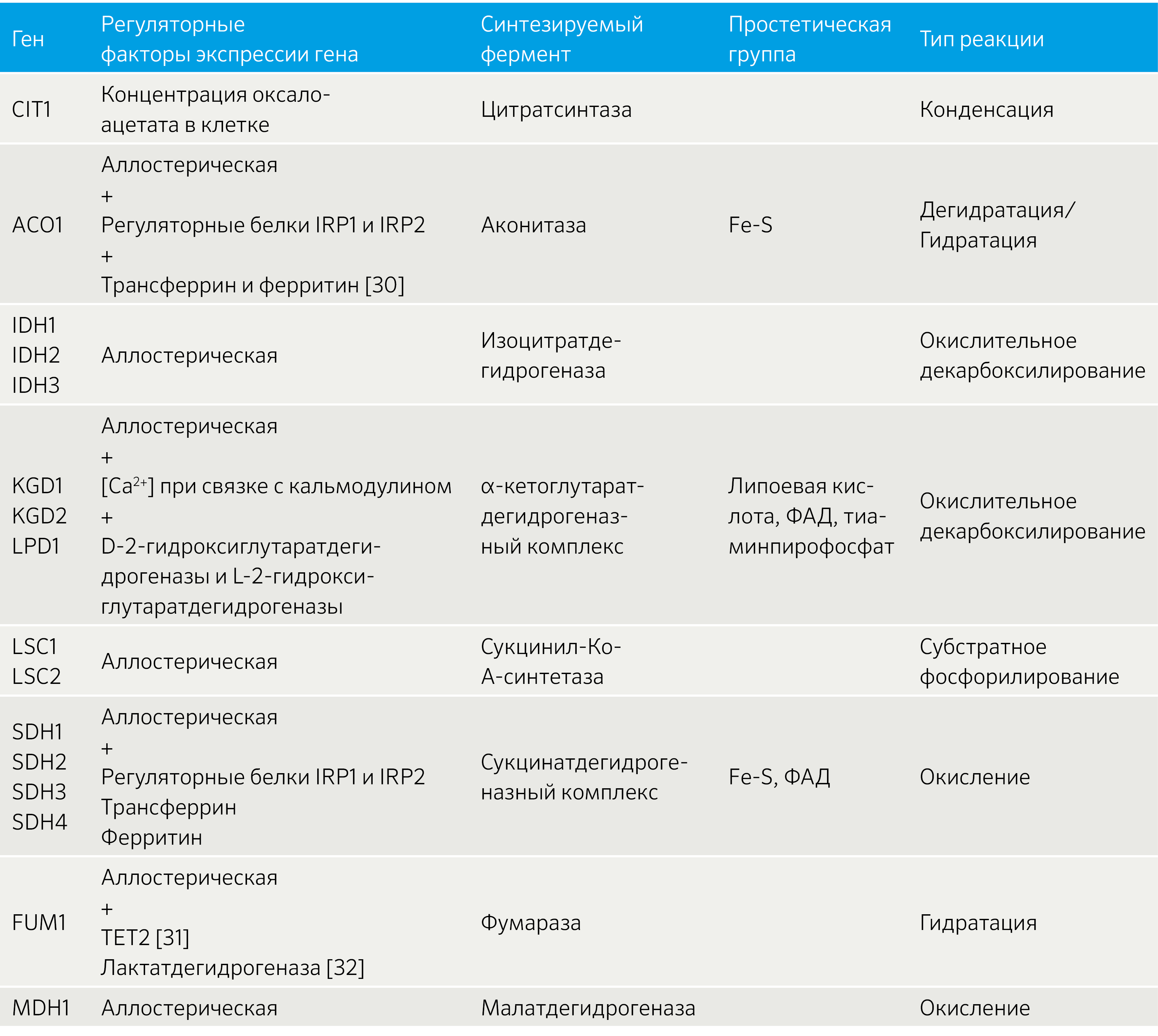
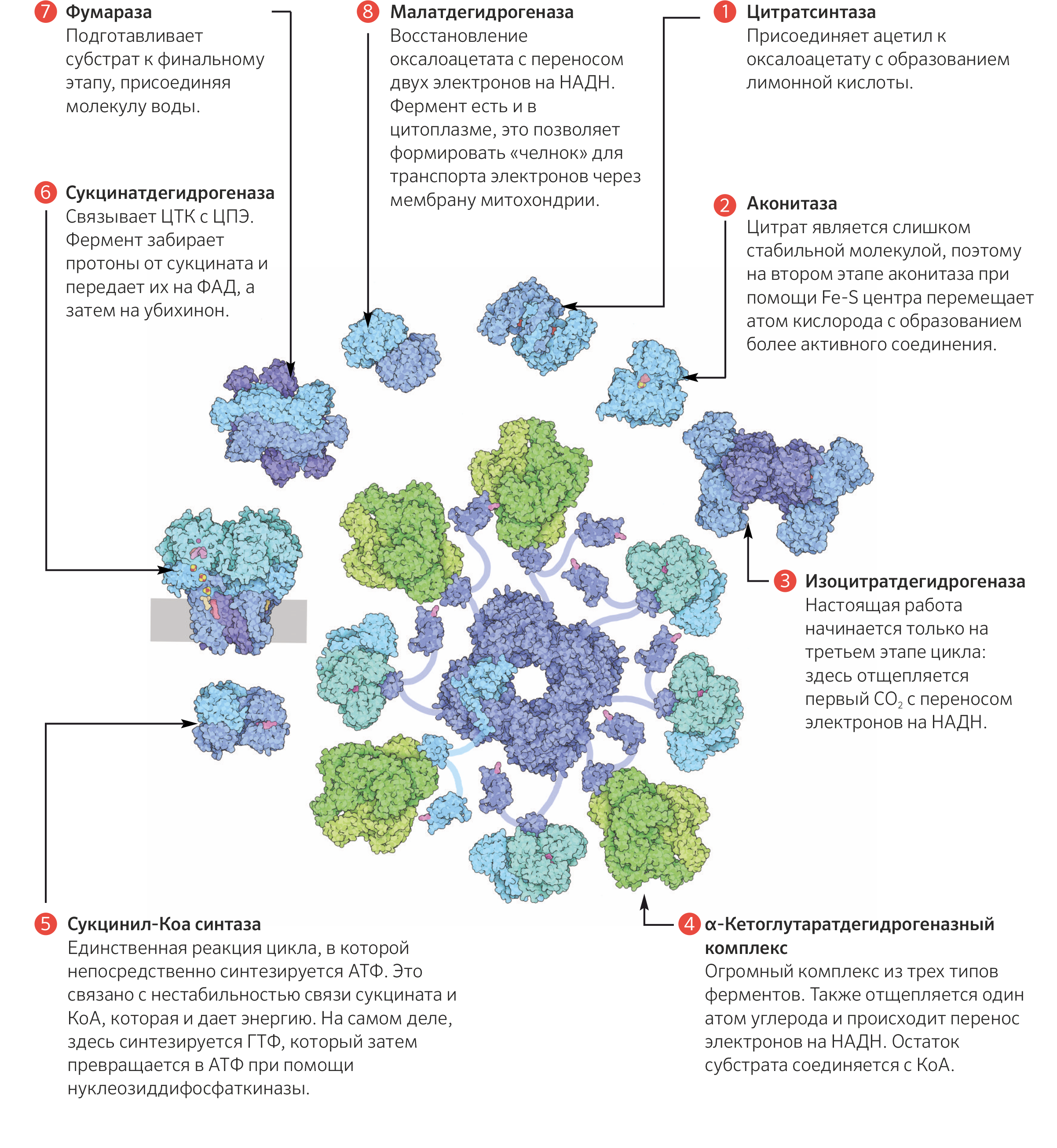
Рисунок 31. Структура ферментов цикла трикарбоновых кислот и их обобщённая роль [33].
КЛИНИЧЕСКОЕ ЗНАЧЕНИЕ ЦТК И ВОЗМОЖНОСТИ ЕГО ФАРМАКОЛОГИЧЕСКОГО КОНТРОЛЯ
Применение сукцината (янтарной кислоты) и витамина B2 в терапии
Применение фармакологических препаратов с целью активации (поддержания, стимуляции) и дезактивации ЦТК довольно дискуссионно и неоднозначно, т. к. сам процесс реакций ЦТК является консолидированным, а факторов, влияющих на реакционную способность, не всегда зависящих друг от друга, а значит — плохо контролируемых, множество. Есть очень примечательные доводы о том, что потребление кислорода кардиомиоцитами вовсе не изменяется по сравнению с изначальным уровнем при селективной блокаде К+-ATФ-зависимых каналов митохондрий, служащих для индукции адаптации к гипоксии, 5-гидроксидеканоатом [34]. То есть многие типовые патологические процессы на самом деле не такие уж и типовые. Очень важно учитывать контекст конкретных факторов, клеток и реакционных процессов.
Нарушение синтеза АТФ, как правило, связано с дезорганизацией дыхательных МФК на фоне патологического состояния (гипоксии). К МФК 1 относят активацию НАД-зависимого окисления субстратов ЦТК, а к МФК 2 активацию сукцинатоксидазного пути окисления (VI стадия ЦТК) как самого энергоэффективного [35]. Применение препаратов янтарной кислоты является спорной практикой. При этом этилметилгидроксипиридина сукцинат (Мексидол, Мексикор, Нейрокс и т. д.) является одним из самых популярных препаратов на догоспитальном, госпитальном и амбулаторном этапах в России [36] и включен в Перечень жизненно необходимых и важнейших лекарственных препаратов [37], но не одобрен для медицинского применения в США и Европе, а также не входит в систему классификации анатомо-терапевтических и химических веществ (Anatomical Therapeutic Chemical Classification System), утвержденную ВОЗ.
В отечественной медицине препарат получил широкое применение при лечении ишемического инсульта [38,39]. Главным доказательным мотивом выступает принцип Ле Шателье-Брауна, т. е. если в ЦТК извне увеличивать концентрацию его субстрата, то в итоге должна получиться вполне естественная стимуляция реакций, не противоречащая законам химии. Но подтверждается ли это эмпирически? Пока нет. Так, исследователи считают увеличение концентрации сукцината не только универсальным диагностическим критерием ишемии миокарда, но и дополнительным фактором повреждения при постинфарктной реперфузии наряду с АФК (рисунок 32) [40].
Накопление сукцината вследствие мутаций по генам сукцинатдегидрогеназы связывают с ингибированием ряда α-кетоглутарат-зависимых гистон-деметилаз, в частности Jhd1. Предполагается, что сукцинат может играть роль в злокачественной трансформации клеток [41]. В итоге терапия в подобных случаях должна быть направлена на ингибирование накопления или чрезмерного окисления сукцината. На сегодняшний день есть данные о том, что вещество диметилмалонат обладает кардиопротективным свойством на экспериментальных моделях за счет воздействия на процесс окисления сукцината [42]. Правда, такие данные должны подтверждаться надлежащими научными исследованиями с участием людей, т. к. клиническое значение такого подхода пока тоже не выяснено или отсутствует вовсе.
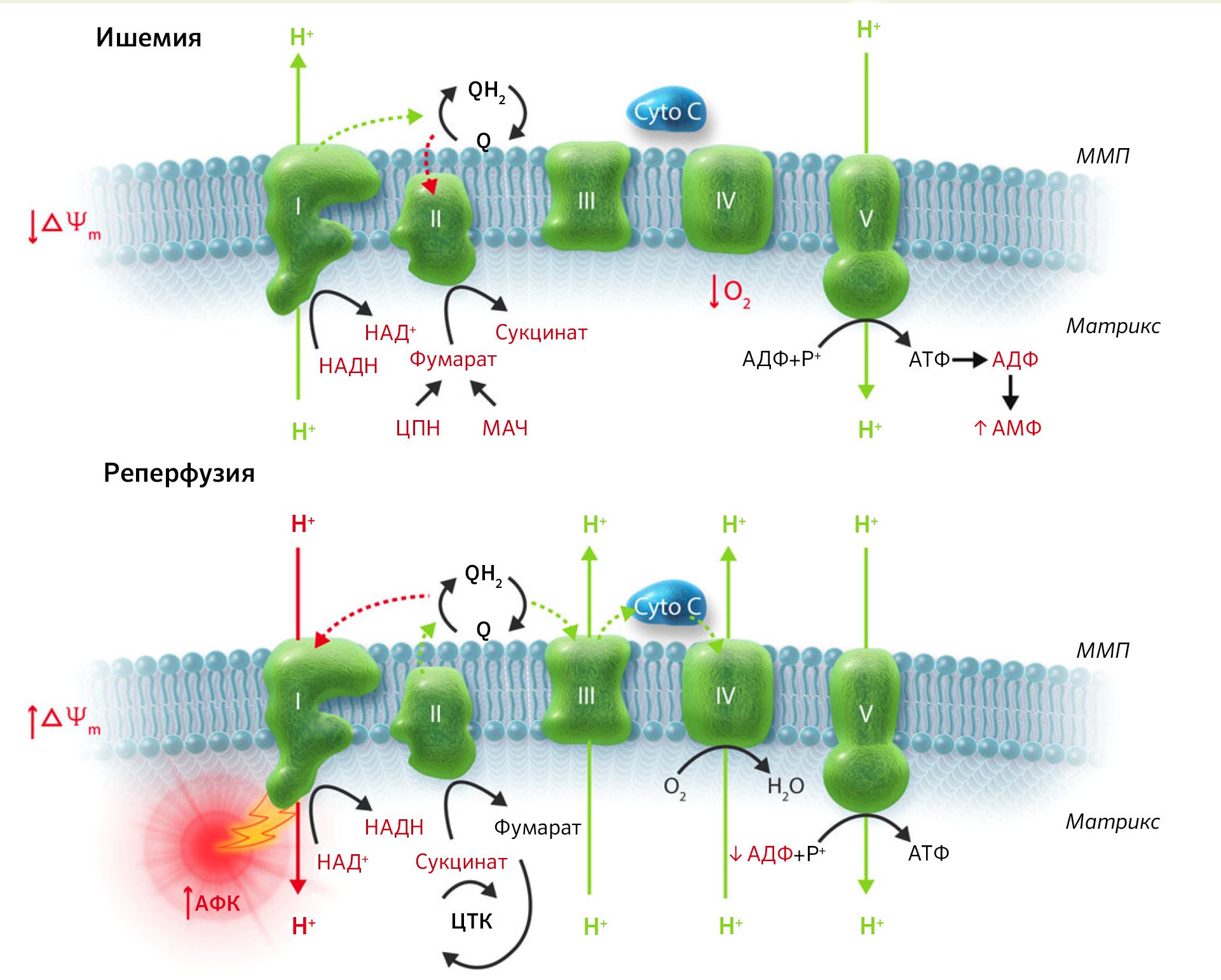
В любом случае нужно понимать, что есть биохимия in silico\in vitro\in vivo, но в противовес есть парадигма доказательной медицины и клиническое значение для практики врача.
Касаемо рибофлавина, во-первых, выше было сказано, что витамин В2 является предшественником кофермента ФАД в организме человека. Поэтому будет закономерно рассматривать влияние рибофлавина с позиции метаболизма ФАД. Во-вторых, окисление пирувата, α-кетоглутарата и сукцината идет под действием дегидрогеназных комплексов, обязательным компонентом которых является ФАД. В третьих, ФАД обладает высоким окислительно-восстановительным потенциалом (больше, чем НАД+ при значении Е° –0,32 против –0,12 у ФАД). Опираясь на предыдущие доводы касательно нежелательного накопления сукцината, есть вероятность его опосредованной утилизации путем потенцирования окисления до фумарата. Но есть загвоздка: реакция превращения сукцината в фумарат обратима, а сукцинатдегидрогеназный комплекс, как и любой фермент, может ускорить как прямую, так и обратную реакции. В таком случае направление реакции будет диктоваться концентрацией субстрата или продукта, но контролировать и тем более влиять на это в биосистеме человека пока непосильная задача. И может быть не нужная.
Все же ФАД (и НАД+) имеют некоторое диагностическое значение, например, как мишени при флуоресценции (хемилюминесценции) слизистых ротовой полости при обследовании онкологических больных [43,44]. Дело в том, что восстановленные формы этих коферментов могут поглощать спектр излучения в 340 нм, а окисленные нет. В итоге есть возможность качественно регистрировать интенсивность окисления, особенно при усилении хемилюминесценции от АФК [45].
Вдобавок необходимо обозначить, что дефицит рибофлавина не так распространен, а последствия его недостатка не настолько опасны, чтобы употреблять его в виде препарата. Ведь фактическую суточную норму жители экономически благополучных стран получают с пищей, и вопрос скорее стоит в сбалансированности питания.
Соответственно трендом и перспективой на сегодня является не использование компонентов ЦТК для моделирования реакций при метаболических патологиях любого генеза или применение их в виде препаратов для лечения, а скорее их прикладное исследование в клинической лабораторной диагностике. Например, жидкостная хроматография с масс-спектрометрией для определения онкометаболита D-2-гидроксиглутарата, о котором будет сказано ниже, или газовая хроматография с масс-спектрометрией для определения концентрации сукцината и фумарата.
Онкология, митохондриальные заболевания и иммунитет
Ферменты ЦТК кодируются конститутивными генами (housekeeping genes). Звучит скучно, если это не такие заболевания, как лейомиома или феохромоцитома, возникающие из-за дефекта в гене, кодирующем сукцинатдегидрогеназный комплекс (SDHB, SDHC, SDHD [46]), а также синдромы Лея, Кернса-Сейра, MELAS, MERRF [47]. Некоторые мутации в гене FUM1, кодирующем фумаразу, приводят к снижению или потере активности фермента и как следствие образованию опухолей кожи, матки, нейробластом и/или рака почек (например, лейомиоматоз и хромофильная карцинома почек второго типа) [48]. Для митохондриальных заболеваний характерна гетероплазмия, т. е. в клетке могут находиться как мутантные мтДНК, так и ДНК дикого типа [49]. Также появились данные, что мтДНК может наследоваться потомками от отца [50].
Давно известно, что пусковым механизмом онкологических процессов является собственное снижение реакционной способности ЦТК при нарушениях в ЦПЭ ввиду деградации ферментов, а также концентрирование онкогенных метаболитов [51]. Например, при мутациях в генах IDH1 и IDH2 изоцитратдегидрогеназы образуется субстрат D-2-гидроксиглутарат [52]. В ответ на накопление данного субстрата экспрессируется белок HIF-1α (Hypoxia-inducible factor 1-alpha — фактор, индуцируемый гипоксией), являющийся онкомаркером, т. к. способствует ангиогенезу за счет взаимодействия с проангиогенным фактором VEGF (Vascular Endothelial Growth Factor — фактор роста эндотелия сосудов) [53].
Гипоксию не стоит рассматривать только с позиции физического снижения концентрации кислорода ввиду нарушения его поступления в кровь или транспорта в ткани. Гипоксия также может быть сигналом нарушения тканевого дыхания (функции митохондрии), неотъемлемой частью которого является ЦТК, и об этом было сказано в самом начале. Особую ее роль может подтверждать тот факт, что нейтрофилы в условиях недостатка кислорода живут значительно дольше, чем в нормальных. Такая живучесть определяется увеличением количества транскрипционных факторов HIF-1α, которые приводят к экспрессии Mcl-1 (Induced myeloid leukemia cell differentiation protein — индуцированный белок дифференцировки клеток миелоидного лейкоза) — главного антиапоптотического фактора нейтрофилов из семейства Bcl-2 [54,55].
Не исключают и эффект Варбурга, который сопряжен с выключением митохондриальных генов и сопутствует онкологическим процессам [56]. Данный эффект основан на активации интенсивного гликолиза с образованием лактата даже при избытке кислорода, что является облигатным признаком пролиферации опухолевых клеток [57,58]. При этом некоторые стадии ЦТК обращаются, чтобы синтезировать ацетил-КоА, который нужен для синтеза липидов. Также HIF-1α действует как фактор транскрипции, усиливающий синтез гликолитических ферментов, киназы пируватдегидрогеназного комплекса, которая блокирует реакцию окислительного декарбоксилирования пирувата, и тем самым возрастает концентрация пирувата в цитоплазме клетки, что приводит к усилению эффекта Варбурга и метаболизированию пирувата в лактат.
В конечном итоге при накоплении D-2-гидроксиглутарата возрастает риск сопряженного острого миелоидного лейкоза или глиомы [59,60], при сопутствующем нарушении деметилирования ДНК с участием эпигеномного фактора TET2 (Ten-eleven Translation\Tet methylcytosine dioxygenase 2 — тетраметилцитозиндиоксигеназа 2) [61]. Уменьшение количества HIF-1α происходит при действии белка VHL (Von Hippel–Lindau tumor suppressor protein — белок-онкосупрессор), но не в условиях гипоксии (62).
Синдром Лея — генетически детерминированное заболевание с поражением серого вещества головного и спинного мозга, чаще проявляется в раннем детском возрасте [63]. МФК транслируются на митохондриальной ДНК, а мутации при синдроме Лея приводят к дисфункции ЦПЭ (особенно МФК 1, 2 и 4), нарушению синтеза пируватдегидрогеназного комплекса с накоплением в дальнейшем пирувата и лактата в тканях [64,65]. Накапливаемый пируват не поступает в митохондрии, и образующийся в чрезмерных концентрациях лактат оказывает токсичное действие на структуру нейрона.
Синдром Кернса-Сейра или митохондриальная миопатия имеет характерный симптомокомплекс: прогрессирующая наружная офтальмоплегия, пигментная ретинопатия, атриовентрикулярная блокада. Наиболее часто патология выражена со стороны глаз в виде птоза. При этом заболевании также наблюдаются нарушения в работе эндокринной системы. Они включают гипогонадизм, сахарный диабет, гипопаратиреоз, дефицит соматотропина [66]. Синдром проявляется при делеции митохондриальной ДНК [67], что приводит к гетероплазмии [68], и уже распределение мутантных ДНК зависит от локализации процесса в определенной клетке, ткани, органе [69]. Мутации, как правило, приводят к выключению МФК ЦПЭ, что создает уменьшение соотношения [АТФ]/[АДФ], аллостерической деградации реакционной способности ЦТК и накоплению пирувата и лактата [70].
Синдром MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды) — нейродегенеративное заболевание, диагностически значимыми жалобами которого являются мигрени, судороги, снижение слуха, непереносимость физических нагрузок и повышение концентрации лактата в крови [71].
MERRF (англ. Myoclonic Epilepsy with Ragged Red Fibers) — заболевание, характеризующиеся миоклонической эпилепсией и феноменом RRF (морфологический маркер в биоптате миофибрилл с рваными красными краями). Патогенез данного синдрома является похожим на вышеизложенные митохондриальные заболевания [72].
Иммунная система и ее вовлеченность при нарушениях синтеза ферментов ЦТК понятна. Очевидно, что клетки, потерявшие функциональность своих митохондрий, заведомо являются чужеродными и провоцирующими иммунный ответ. А что можно сказать о прямом воздействии на ЦТК самих иммунокомпетентных клеток?
Существует мнение, что гамма-интерферон не только активирует макрофаги и NK-клетки для регуляции аутоиммунных процессов, но и потенцирует аэробный гликолиз, перепрофилирует митохондрии, усиливает сигналинг окислительного фосфорилирования в них, но это лишь опосредованное влияние на ЦТК как на реакционный компонент митохондрий [73]. В международной практике иммуностимуляторами называют и сами цитокины, используемые как фармакологические средства, которые одобрило FDA. Это сигнальные молекулы иммунитета, и они взаимодействуют с мембранными рецепторами клеток, но не митохондрий.
Тот же интерферон не взаимодействует напрямую с рецепторами митохондрий. Внутриклеточные АФК, сопровождающие воспаление (хотя определенные концентрации АФК стабильны и не являются токсичными, например, перекись водорода присутствует в клетке в постоянной концентрации в пределах 1 нМ [74]), формируются за счет связи интерферона с цитоплазматическим рецептором врожденного иммунитета NLRX1 и адаптерного белка MAVS на внешней мембране митохондрий. Тем самым MAVS, при наличии митохондриальных АФК, может участвовать в апоптозе зараженных вирусами клеток или нетозе нейтрофилов [75,76,77]. Но нет утверждения, что прием определенного интерферона или его индуктора в виде препарата может достоверно повлиять на энергетический метаболизм пораженных вирусом или иммунокомпетентных клеток, тем самым усиливая их активность. На сегодня не продемонстрирована такая эффективность в рамках двойных слепых плацебо-контролируемых исследований, поэтому использование иммуностимуляторов считается бесполезным. Перспектива исследований скорее затрагивает влияние на сигналинг в ЦПЭ, так как ЦТК является последовательностью реакций небелковых компонентов, находящихся в зависимости от функционирования огромного комплекса белкового сигналинга митохондрий. Однако исследователями показано возрастание L-2-гидроксиглутарата (при все той же мутации в генах изоцитратдегидрогеназы) и его производных в ответ на гипоксию и экспрессию HIF-1α у CD8+ лимфоцитов [78].
Таким образом, метаболиты, выявляющиеся при нарушении работы ЦТК и митохондрий, в целом, могут являться как токсичными (лактат, АФК), так и онкогенными (VEGF, D-2-гидроксиглутарат, HIF-1α). Но нет оснований полагать, что ЦТК можно контролировать и регулировать клинико-фармакологическими методами.
СПИСОК ЛИТЕРАТУРЫ
- Sagan L. On the origin of mitosing cells. Journal of Theoretical Biology. – 1967. – Vol. 14. – №3. – P. 225–226. doi:10.1016/0022-5193(67)90079-3
- Evans M.C. et al. A new ferredoxin-dependent carbon reduction cycle in a photosynthetic bacterium / Evans M.C. Buchanan B.B. Arnon D.I // Proc Natl Acad Sci USA. – 1966. – Vol. 55. – №4. – P. 928–934.
- Smith E. et al. Universality in intermediary metabolism. / Smith E. Morowitz H. // Proc Natl Acad Sci USA. – 2004. – Vol.101. – №36. – P. 13168–13173.
- Klatt C.G. et al. Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. / Klatt C.G. Bryant D.A. Ward D.M. // Environ Microbiol. – 2007. – Vol. 9. – №8. – P. 2067– 2078.
- Julian J. et al. Photosynthesis: Plastid Biology, Energy Conversion and Carbon Assimilation. /Julian J. Eaton-Rye. Baishnab C. Tripathy. Thomas D. Sharkey. // – 2012. – P. 654– 856.
- The Nobel Prize in Physiology or Medicine 1937. The Nobel Foundation. https://www.nobelprize.org/pri...
- The Nobel Prize in Physiology or Medicine 1953. The Nobel Foundation. https://www.nobelprize.org/pri...
- McCammon M. T. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism: isolation and characterization of Acn— mutants .Genetics. – 1996. – Vol. 144. – P. 57–69.
- Udvardi M.K. et al. Isolation and characterization of a cDNA encoding NADP+-specific isocitrate dehydrogenase from soybean (Glycine max)./Udvardi M.K. Mc Dermott T.R. Kahn M.L// Plant Mol Biol – 1993. – Vol. 21. P. 739.
- Repetto B. et al. In vivo assembly of yeast mitochondrial alpha-ketoglutarate dehydrogenase complex./Repetto B. Tzagoloff A.// Mol Cell Biol. –1991. – №8.– P. 3931 – 3939.
- Nishimura J.S. et al. Isolation, amino acid analyses and refolding of subunits of pig heart succinyl-CoA synthetase./Nishimura J.S. Ybarra J. Mitchell T. Horowitz P.M.// Biochem J. – 1988. – Vol. 250. – № 2.– P. 429 – 434.
- Chapman K.B. et al. SDH1, the gene encoding the succinate dehydrogenase flavoprotein subunit from Saccharomyces cerevisiae. /Chapman K.B. Solomon S.D. Boeke J.D.//Gene. – 1992. – №1. – P. 131 –136.
- Lombardo A. et al. Cloning and characterization of the iron-sulfur subunit gene of succinate dehydrogenase from Saccharomyces cerevisiae. /Lombardo A. Carine K. Scheffler I.E.//J Biol Chem. – 1990 .– Vol. 265.– № 18. – P. 0419 –10423.
- Bullis B.L. et al. Isolation and characterization of the Saccharomyces cerevisiae SDH4 gene encoding a membrane anchor subunit of succinate dehydrogenase. /Bullis B.L. Lemire B.D.//J Biol Chem. – 1994. – Vol. 269.– №9 – P. 6543–6549.
- Schägger H. et al. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form./ Schägger H. von Jagow G.//Anal Biochem. – 1991. – №2. – P. 223– 231.
- Dibrov E. et al. The Saccharomyces cerevisiae TCM62 gene encodes a chaperone necessary for the assembly of the mitochondrial succinate dehydrogenase (complex II). /Dibrov E. Fu S. Lemire B.D.//J Biol Chem. – 1998. – Vol. 273. – № 48 – P. 32042 – 32048.
- Peleg Y. et al. Inducible overexpression of the FUM1 gene in Saccharomyces cerevisiae: localization of fumarase and efficient fumaric acid bioconversion to L-malic acid./Peleg Y. Rokem J.S. Goldberg I. Pines O.// Appl Environ Microbiol. – 1990.– Vol. 56. – №9. – P. 2777–2783.
- Bock R.M. et al . (1953). Studies of the Enzyme Fumarase. I. Kinetics and Equilibrium. /Bock. R. M. & Alberty. R. A.// Journal of the American Chemical Society. –1953. – Vol. 75. – №8. – P. 1921–1925.
- Genda Т. et al. Purification and characterization of fumarase from Corynebacterium glutamicum./Genda Т. Watabe S. Ozaki Hy.// Biosci. Biotechnol. Biochem. – 2006.– Vol. 70. – №5. – P. 1102–1109.
- McAlister-Henn L. et al. Isolation and expression of the gene encoding yeast mitochondrial malate dehydrogenase./McAlister-Henn L. Thompson L.M.// J Bacteriol. – 1987.– Vol. 169. – №11. – P. 5157–5166.
- Boyer P.D. The ATP synthase--a splendid molecular machine. Annu Rev Biochem. –1997.– №66. – P. 717–749.
- Lancaster E.M. et al. Acetaminophen hepatotoxicity: an updated review./Lancaster E.M. Hiatt J.R. Zarrinpar A.// Arch Toxicol.– 2015. – Vol. 89. – №2.– P. 193–199. doi: 10.1007/s00204–014–1432–2.
- Jaeschke H. et al. Intracellular signaling mechanisms of acetaminophen-induced liver cell death./Jaeschke H. Bajt M.L.// Toxicol Sci. – 2006.– Vol. 89. – P. 31–41.
- Walker J.E. et al. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold./Walker J.E. Saraste M. Runswick M.J. Gay N.J.// EMBO J.– 1982. – Vol. 1. – №8. – P. 945–951.
- Pfanner N. et ll. Mitochondrial proteins: from biogenesis to functional networks./Pfanner N. Warscheid B. Wiedemann N.// Nat Rev Mol Cell Biol. – 2019.
- Adler V. et al. Role of redox potential and reactive oxygen species in stress signaling. /Adler. V. Yin. Z.// Oncogene. – 1999. – Vol. 18 .– №45. – P. 6104–6111
- Itoh K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain / Itoh. K. Wakabayashi. N.// Genes Dev. – 1999. – Vol. 13 .– №1. – P. 76– 86
- Анастасина М.С. и соавт. Цикл Кребса: транскрипционная регуляция генов у дрожжей и Митохондриальные заболевания человека. /Анастасина М.С. Самбук Е.В. // Biological Communications. – 2009. – №2. С. 40– 41
- Liu Z. et al. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function./Liu. Z. and Butow. R. A.// Molecular and Cellular Biology. – 1999. – Vol. 19. – № 10 – P. 6720–6728.
- Rouault T.A. et al. Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications./Rouault T.A. Stout C.D. Kaptain S. Harford J.B. Klausner R.D// Cell. – 1991. – Vol. 64 – P. 881–883.
- Xiao M. et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumour suppressors./Xiao M. Yang . Xu W. Ma S. Lin H. Zhu H. Liu L. Liu Y. Yang C. Xu Y. Zhao S. Ye D. Xiong Y and Guan K.L .// Genes Dev. –2012. – Vol. 26. – P. 1326–1338
- Hamanaka R.B. et al. Targeting glucose metabolism for cancer therapy. /Hamanaka R.B. Chandel N.S.//J Exp Med. – 2012. – Vol. 209. – №2. – P. 211–215.
- Voet D. et al.Fundamentals of Biochemistry. /Voet. D. Voet. J.G. Pratt.C.W.// John Wiley & Sons Inc.: New York, NY, USA. –2002.
- Chen Y. et al. Selective blockade of mitochondrial K(ATP) channels does not impair myocardial oxygen consumption. /Chen Y. Traverse J.H. Zhang J. Bache R.J.// Am J Physiol Heart Circ Physiol. – 2001. – Vol. 281. – №2. – P. 738–744.
- Тихонова Е.О. и соавт . Использование препаратов, содержащих сукцинат, в клинике инфекционных болезней./Тихонова Е.О. Ляпина Е.П. Шульдяков А.А. Сатарова С.А. // Терапевтический архив.– 2016. – Vol. 88. – №11. – P. 121–122
- Likhacheva E.B. et al. [Clinical and immunological assessment of efficacy of mexidol in the treatment of lumbosacral radiculopathy]./Likhacheva E.B. Sholomov I.I.// Zh Nevrol Psikhiatr Im S S Korsakova. – 2006. – Vol. 106. – №10. – P. 52–57.
- Об утверждении перечней жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2016 год. Распоряжение от 26 декабря 2015 года №2724–р. [On approval of the list of vital and essential medicinal products for medical use by 2016. The order from December 26, 2015 No. 2724–p]. http://government.ru/docs/2136...
- Громова О.А. и соавт . Хемореактомный анализ сукцината этилметилгидроксипиридина./Громова О.А. Торшин И.Ю. Федотова Л.Э. Громов А.Н. // Неврология, нейропсихиатрия, психосоматика. – 2016. – №3. – С. 54
- Завалий Л.Б. и соавт. МЕТАБОЛИЧЕСКАЯ ТЕРАПИЯ ПРИ ИШЕМИЧЕСКОМ ИНСУЛЬТЕ. /Завалий Л.Б. Петриков С.С. Щеголев А.В.// НМП. – 2018. – №1.
- Victoria R. et al. Succinate metabolism: a new therapeutic target for myocardial reperfusion injury./Victoria R. Pell. Edward T. Chouchani. Christian Frezza. Michael P. Murphy. Thomas Krieg.// Cardiovascular Research. – 2016. – Vol. 111. – №2, – P. 134–141
- Smith E.H. et al. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma./Smith E.H. Janknecht R. Maher L.J.// Hum Mol Genet. – 2007. – Vol. 16. – №24. – P. 3136–3148.
- Boylston J.A. et al. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury./ Boylston J.A. Sun J. Chen Y. Gucek M. Sack M.N. Murphy E.// J Mol Cell Cardiol. – 2015. – Vol. 88 – №73. – P. 81.
- Huang T.T. et al. Two-channel autofluorescence analysis for oral cancer./ Huang T.T. Chen K.C. Wong T.Y. Chen C.Y. Chen W.C. Chen Y.C. Chang M.H. Wu D.Y. Huang T.Y. Nioka S. Chung P.C. Huang J.S.// J Biomed Opt. – 2018. – Vol. 24. – №5. – P. 1–10.
- Massey V. et al . The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem./Massey V. Strickland S. Mayhew S.G. Howell L.G. Engel P.C. Matthews R.G. Schuman M. Sullivan P.A. // Biophys.Res. Commun. – 1969. – Vol. 36. – №6. – P. 891–897.
- Faulkner K. et al. Luminol and lucigenin as detectors for O2. / Faulkner K. Fridovich I.// Free Radic. Biol. Med. – 1993. – Vol. 15. – №4. – P. 447–451.
- Astuti D. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma./Astuti D. Latif F. Dallol A. Dahia P. L. Douglas F. George E. Skoelberg F. Husebye E. S. Eng C. Maher E. R. // Am. J. Hum. Genet. – 2001. – Vol. 69. – №1. P. 49–54.
- Rustin P. et al. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. /Rustin P. Bourgeron T. Parfait B. Chretien D. Munnich A. Rötig A.// Biochim Biophys Acta. – 1997. – Vol. 22. – №1361. – P. 185–197.
- Tomlinson I. P. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer./ Tomlinson I. P. Alam N. A. Rowan A. J. Barclay E. Jaeger E. E. Kelsell D. Leigh I. Gorman P. Lamlum H. // Nat. Genet. – 2002. – Vol. 30. – №4. – P. 406–410.
- Gorman G.S. et al. Mitochondrial diseases./Gorman GS. Chinnery P.F. DiMauro S. Hirano M. Koga Y. Mc Farland R. Suomalainen A. Thorburn D.R. Zeviani M. Turnbull D.M.// Nat Rev Dis Primers. – 2016. – Vol. 2.– №16080.
- Shiyu Luo. et al. Atwal, Taosheng Huang Biparental Inheritance of Mitochondrial DNA in Humans./Shiyu Luo. C. Alexander Valencia. Jinglan Zhang. Ni-Chung Lee. Jesse Slone. Baoheng Gui. Xinjian Wang. Zhuo Li. Sarah Dell. Jenice Brown. Stella Maris Chen. Yin-Hsiu Chien. Wuh-Liang Hwu. Pi-Chuan Fan. Lee-Jun Wong. Paldeep S. // Proceedings of the National Academy of Sciences Dec. – 2018. – Vol. 115. – №51. – P. 13039-13044
- Edoardo G. et al. Defects in mitochondrial metabolism and cancer./Edoardo G. Christian F.// Cancer & Metabolism. – 2014 .– Vol. 2. – №1.
- Ward P.S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. / Ward P.S. Patel J. Wise D. witz J.D. Carroll M. Su S.M. Sharp K.A. Levine R.L. Thompson CB.// Cancer Cell. – 2010. – Vol .17. –№3. – P. 225–234.
- Semenza G.L. Targeting HIF-1 for cancer therapy. Nature Reviews Cancer. – 2003.– Vol . 3. – №10. – P. 721–732.
- Walmsley S. R. et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. /Walmsley. S. R. Print. C. Farahi. N. Peyssonnaux. C. Johnson. R. S. Cramer. T. Chilvers. E. R.// The Journal of Experimental Medicine. – 2005. – Vol. 201. – №1. – P. 105–115.
- Piret J. P. et al. Hypoxia-inducible factor–1–dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis./Piret. J. P. Minet. E. Cosse. J. P. Ninane. N. Debacq. C. Raes. M. & Michiels. C.// Journal of Biological Chemistry .– 2005. – Vol. 280. – №10. – P. 9336–9344.
- López-Lázaro M. The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem. – 2008. – Vol. 8. – №3. – P. 305–312.
- Alfarouk K.O. et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. / Alfarouk K.O. Verduzco D. Rauch C. Muddathir A.K. Adil H.H. Elhassan G.O. Ibrahim M.E. David Polo Orozco J. Cardone R.A. Reshkin S.J. Harguindey S. // Oncoscience. – 2014. – Vol. 1. – №12 – P. 777–802.
- Gatenby R.A. et al. Why do cancers have high aerobic glycolysis? /Gatenby R.A. Gillies R.J. // Nat Rev Cancer. – 2004. – Vol. 4. – №11. – P. 891–899.
- Capper D. et al. Monoclonal antibody specific for IDH1 R132H mutation./Capper D. Zentgraf H. Balss J. Hartmann C. von Deimling A. // Acta Neuropathol. – 2009. – Vol. 118. – №5. – P. 599–601.
- Wang Y. et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation./Wang Y. Xiao M. Chen X. Chen L. Xu Y. Lv L. Wang P. Yang H. Ma S. Lin H. Jiao B. Ren R. Ye D. Guan K.L. Xiong Y. // Mol Cell. – 2015. – Vol. 57. – №4. – P. 662–673.
- Heuser M. et al. Epigenetics in myelodysplastic syndromes./Heuser M. Yun H. Thol F. // Semin Cancer Biol. – 2018. – Vol. 51. – P. 170–179.
- Kaelin W.G. The von Hippel–Lindau tumor suppressor protein: roles in cancer and oxygen sensing. Cold Spring Harb Symp Quant Biol. – 2005. – Vol. 70. – P. 159–166.
- Leigh D. Subacute necrotising encephalopathy in an infant. J Neurol Neurosurg Psychiatry – 1951. – Vol. 14 – P. 216–221.
- Finsterer J. Leigh and Leigh–like syndrome in children and adults. Pediatr Neurol. – 2008. – Vol. 39.– №4. – P. 223–235.
- Gerards M. et al. Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options./Gerards M. Sallevelt S.C. Smeets H.J.// Mol Genet Metab. – 2016. – Vol. 117.– №3. – P. 300–312.
- Прыгунова Т.М. и соавт. Митохондриальные заболевания в детской неврологической практике (клиническое наблюдение)./Прыгунова Т.М. Радаева Т.М. Степанова Е.Ю. // Медицинский альманах. – 2014. – Vol. 3. – №33. – С. 85.
- Fine P.E. Mitochondrial inheritance and disease. Lancet. – 1978. – Vol. 2. – №8091. – P. 659–662.
- Chinnery P.F. Modulating heteroplasmy. Trends Genet. – 2002. – Vol. 18. – №4 – P. 173–176.
- Shoubridge E.A. et al. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. /Shoubridge E.A. Johns T. Karpati G.//Hum Mol Genet. – 1997. – Vol. 6. – – №13. – P. 2239–2242.
- Ogasahara S. et al. Improvement of abnormal pyruvate metabolism and cardiac conduction defect with coenzyme Q10 in Kearns-Sayre syndrome./Ogasahara S. Yorifuji S. Nishikawa Y. Takahashi M. Wada K. Hazama T. Nakamura Y. Hashimoto S. Kono N. Tarui S.// Neurology. – 1985. – Vol. 35. – №3. – P. 372–377.
- Pavlakis S.G. et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. /Pavlakis S.G. Phillips P.C. DiMauro S. De Vivo D.C. Rowland L.P.//Ann Neurol. – 1984. – Vol. 16. – №4. – P. 481–488.
- Finsterer J. et al. MERRF Classification: Implications for Diagnosis and Clinical Trials./Finsterer J. Zarrouk-Mahjoub S. Shoffner J.M.// Pediatr Neurol. – 2018. – Vol. 80. – №8. – P. 23.
- Wang F. et al. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. /Wang F. Zhang S. Jeon R. Vuckovic I. Jiang X. Lerman A. Folmes C.D. Dzeja P.D. Herrmann J.// EBioMedicine. – 2018. – Vol. 30. – P. 303–316.
- Hauptmann N. et al. The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. /Hauptmann N. Grimsby J. Shih J.C. Cadenas E.//Arch. Biochem. Biophys. – 1996. – Vol. 335. – №2. – P. 295–304.
- Seth R.B. et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3./Seth R.B. Sun L. Ea C.K. Chen Z.J.// Cell. – 2005. – Vol. 122. – №5. – P. 669.–682.
- Seth R.B. et al. The interferon stimulator mitochondrial antiviral signaling protein facilitates cell death by disrupting the mitochondrial membrane potential and by activating caspases./Seth R.B. Sun L. Ea C.K. Chen Z.J.// J Virol. – 2010. – Vol. 84. – №5. – P. 2421.–2431.
- Arai Y. et al. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation./Arai. Y. Nishinaka. Y. Arai. T. Morita. M. Mizugishi. K. Adachi. S. Takaori-Kondo. A. Watanabe. T. Yamashita. K.// Biochemical and Biophysical Research Communications. – 2014. – Vol. 443. – №2. – P. 556–561.
- Tyrakis P.A. et al. S-2-hydroxyglutarate regulates CD8(+). /Tyrakis P.A. Palazon A. Macias D. Lee K.L. Phan A.T. Veliça P. You J. Chia G.S. Sim J. Doedens A. Abelanet A. Evans C.E. Griffiths J.R. Poellinger L. Goldrath A.W. Johnson R.S. // T-lymphocyte fate. Nature. – 2016. – Vol. 540. – №7632. – P. 236–241.
