
Калиевые каналы внутреннего выпрямления: часть 2

В первой части мы рассмотрели классические калиевые каналы внутреннего выпрямления, которые в первую очередь регулируют возбудимость клеток. Во второй части речь пойдет о подсемействах, участвующих в трансэпителиальном и трансастроцитарном транспорте калия и других ионов. Мутации в этих каналах приводят к дисбалансу электролитов, заболеваниям сетчатки и к сложным неврологическим фенотипам.
Транспортные каналы Kir1.1, 4.x, 5.1 и 7.1
Kir1.1
Kir1.1/KCNJ1 представлен шестью различными сплайс-изоформами и образует гомотетрамеры c проводимостью около 35 пСм [1, 2].
Kir1.1 чувствительны к внутриклеточному (но не к внеклеточному) pH. Закисление цитозоля ведет к закрытию этих каналов с pKa около 6,5 [3–8]. Мутации, сдвигающие pKa к более щелочным значениям, ведут к развитию синдрома Барттера [9–11] (см. раздел «синдром Барттера»).
На COOH-конце субъединицы Kir1.1 есть сигнал локализации в ЭПР (Arg-X-Arg [12]), и, чтобы канал достиг плазматической мембраны, требуется фосфорилирование серин-треонин киназами. Экспрессия Kir1.1 на мембране стимулируется фосфорилированием PKA, SGK-1 и PKC [13] и ингибируется различными WNK (with no lysine/K kinase) киназами. Мутации в WNK-1 и 4 ведут к псевдогипоальдостеронизму II типа (также называется синдромом Гордона). Для пациентов с этим синдромом характерен низкий рост, интеллектуальные нарушения, патологии зубной системы, слабость мышц, тяжелая гипертензия, низкий уровень ренина и альдостерона, метаболический ацидоз, гиперхлоремия и гиперкалиемия. Эффекты измененных вариантов WNK киназ могут быть опосредованы неправильной модуляцией Kir1.1.Так, мутация в WNK-4 снимает ингибирование NCC (Na-Cl cotransporter) и усиливает реабсорбцию NaCl, после чего, вследствие недостатка NaCl в кортикальной части собирательной трубочки, тормозится экскреция K+ [14].
Kir4.x и Kir5.1
Kir4.1/KCNJ10 был одновременно клонирован из кДНК библиотек мозга несколькими группами и описан как KAB-2 [15], BIRK-1 [16], Kir1.2 [17] и BIR10 [18, 19], в той же работе Kir5.1/KCNJ16 описали как BIR9.
Kir4.1 экспрессируется в основном в глиальных клетках [15], на основании чего было сделано предположение о роли этой изоформы в создании пространственного буфера для калия в глиальных клетках (англ. «K+-buffering») [20].
В тканях человека Kir4.1 может экспрессироваться в одиночку, образуя функциональные гомотетрамеры [21], однако он также может гетеромеризоваться с Kir5.1. У таких каналов иные биофизические характеристики, чем у гомомеров Kir4.1: они обладают большей проводимостью отдельных каналов [15, 21–24], более выраженным внутренним выпрямлением (за счет N161 в D/N сайте Kir5.1) и высокой чувствительностью к внутриклеточному pH, поскольку ток через гетеромерные каналы ингибируется при малейшем закислении и усиливается при повышении pH с pKa около 7.5 [22, 24]. Гомомеры Kir5.1 локализуются в цитозоле и не способны формировать функциональные каналы на плазматической мембране.
Kir4.2/KCNJ15 был выделен из кДНК библиотеки человеческой почки [17], а затем другая группа ученых обнаружила этот ген при секвенировании области q22.2 на 21 хромосоме [25]. На COOH-конце Kir4.1, в отличие от Kir4.2 [26], находится АТФ-связывающий мотив Walker A [15]. Как и Kir4.1, Kir4.2 может экспрессироваться как отдельно, так и совместно с Kir5.1 [26, 27].
Kir7.1
Kir7.1/KCNJ13 был независимо обнаружен тремя исследовательскими коллективами [28–30]. Эта изоформа значительно отличается от других каналов семейства своей аминокислотной последовательностью (гомология с ближайшим к нему Kir4.2 составляет всего около 38 %).
Свойства поры Kir7.1 весьма необычны для этого семейства: проводимость отдельных каналов необычайно мала, всего около 50 фСм; канал малочувствителен к Ba2+ и Cs+ (IC50 около 1 и 10 мМ, соответственно); выпрямление тока не зависит от [Mg2+]i, а амплитуда — от [K+]o [28, 29]. За все эти свойства отвечает один аминокислотный остаток M125 (во всех других каналах данного семейства в этой позиции стоит аргинин). Замена метионина на аргинин увеличивает проводимость отдельных каналов приблизительно в 20 раз, а чувствительность к Ba2+ в 10 раз, и выпрямление тока становится таким же, как и у других членов семейства [28, 29].
Как и другие члены семейства, Kir7.1 активируется PIP2, однако аффинность связывания ниже, чем для Kir2.1 [31].
Kir7.1 чувствителен к внутриклеточному pH, причем ток достигает максимальной амплитуды при pH 7.0 и снижается при закислении или защелачивании среды. За такое поведение отвечает остаток гистидина His26 на N-конце белка, который находится в цитоплазме. Мутации этого остатка приводят к повышению активности канала при щелочных значениях pH и усилению ингибирования в кислой среде [32].
Физиологическая роль транспортных каналов Kir
Kir1.1, Kir4.1 и Kir5.1 в почках
Для поддержания объема тканевой жидкости и ионного гомеостаза в организме, в почечных канальцах работают многокомпонентные транспортные системы. Эти системы обладают сложной пространственной организацией: различные каналы и транспортеры экспрессируются в различных отделах нефрона, в различных типах клеток в рамках одного отдела, и даже на апикальной и базолатеральной стороне отдельных клеток. Калиевые каналы необходимы не только для поддержания [K+] в организме, но и для регуляции концентраций Na+ и Cl−. Kir1.1 играет ключевую роль в этом процессе. Он экспрессируется в толстом отделе восходящего колена петли Генле, в дистальном извитом канальце и в собирательной трубочке и всегда располагается на апикальной мембране эпителия [33–35]. В различных отделах нефрона представлены разные изоформы, образующие каналы с различной проводимостью: в толстом отделе восходящего колена петли Генле присутствуют каналы с проводимостью 35 пСм и 70 пСм, в собирательной трубочке проводимость отдельных каналов составляет 35 пСм [36, 37].
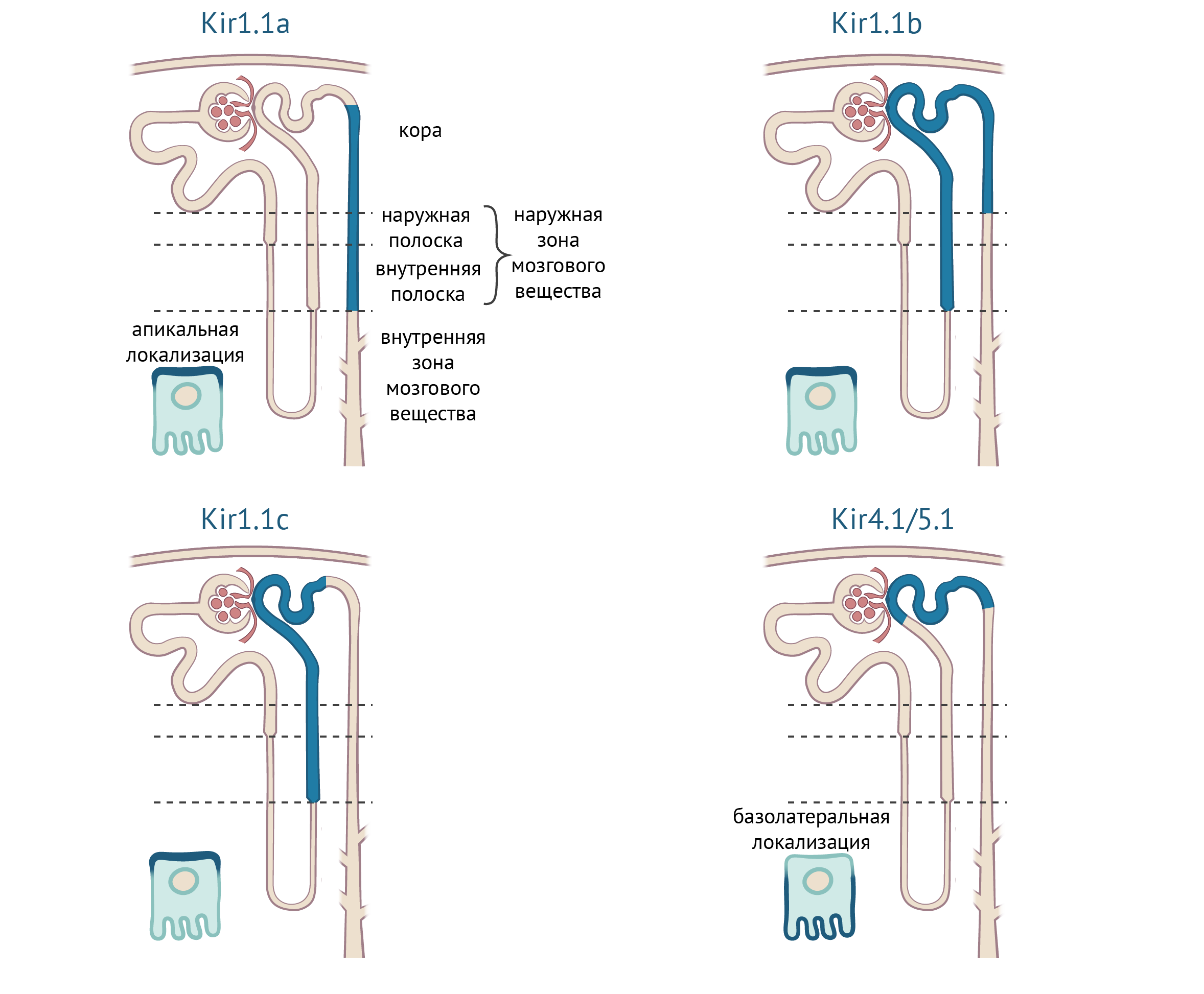
Рисунок 11. Экспрессия транспортных каналов Kir в различных отделах нефрона и их субклеточная локализация. По [19].
В толстом восходящем колене петли Генле реабсорбируется около 25 % ионов натрия в основном за счет транспортера NKCC2 (Na+/K+/2Cl− котранспортер) в апикальной мембране нефроцитов. Этот транспортер, как и нейрональный NKCC1, переносит по одному иону Na+ и K+ и два иона Cl− внутрь клетки, а Kir1.1 поставляет K+ из цитоплазмы к внеклеточному сайту связывания. Этот процесс называется рециклизацией K+ (англ. K+ recycling) [38]. Накопленный в клетке Na+ переносится в интерстициальную жидкость благодаря Na+/K+-АТФазе, расположенной в базолатеральной мембране эпителия. Кроме того, Kir1.1 гиперполяризует мембрану и способствует транспорту Cl− из клетки через базолатеральные хлоридные каналы [38]. Это обеспечивает однонаправленный транспорт Na+ и Cl− через эпителий, а также поддерживает трансэпителиальный потенциал: положительные заряды сконцентрированы в просвете канала (люмене), а отрицательные — в интерстициальной жидкости. Этот потенциал способствует транспорту Na+, Ca2+ и Mg2+ по парацеллюлярному пути [38].
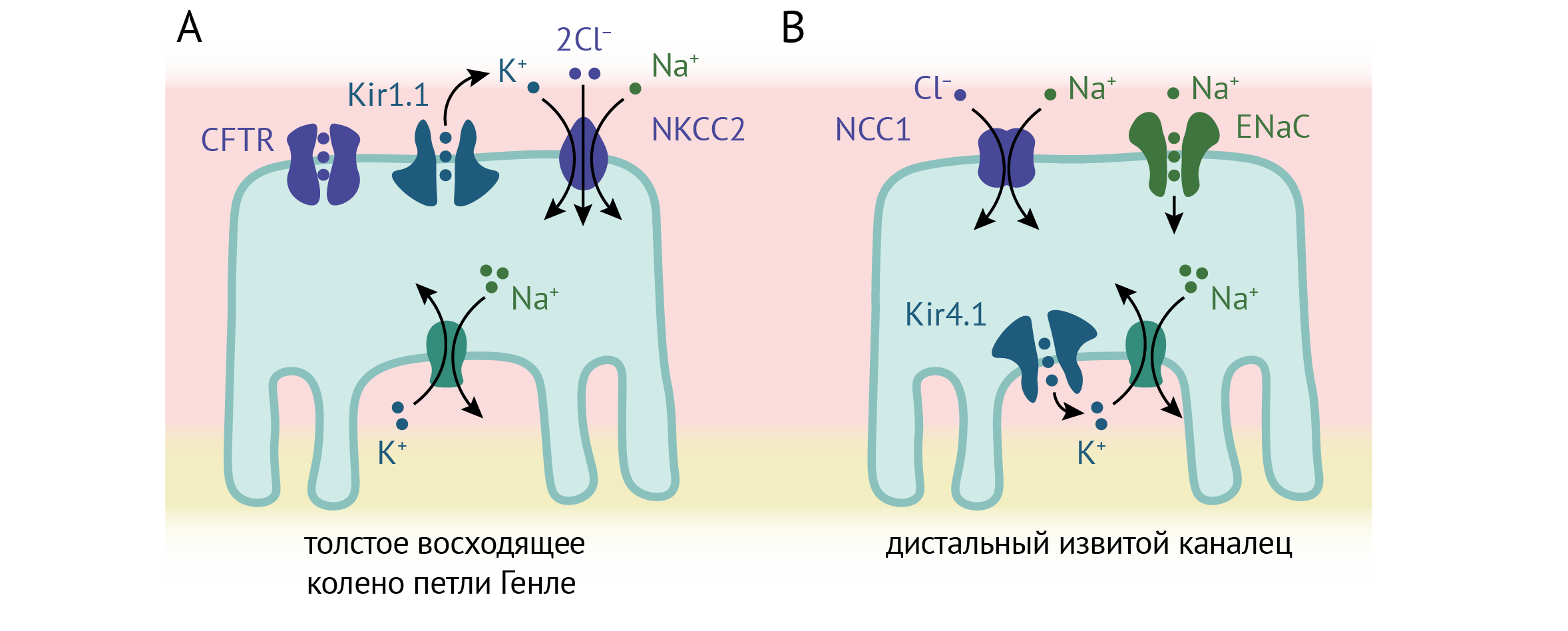
Рисунок 12. Роль транспортных каналов Kir в толстом восходящем отделе колене петли Генле и в дистальном извитом канальце. По [19].
Роль Kir1.1 в рециклизации K+ становится ясной при нарушении работы Kir1.1: у мышей, нокаутных по этому гену, а также у пациентов с синдромом Барттера нарушена реабсорбция NaCl [36, 37].
Kir1.1 в апикальной мембране функционально связан с Cl− каналом CFTR (cystic fibrosis transmembrane regulator — трансмембранный регулятор муковисцидоза) [39], который уменьшает активность Kir1.1. Это взаимодействие может быть вовлечено в реализацию эффектов антидиуретического гормона, который увеличивает проводимость Kir1.1 в дистальном извитом канальце и собирательной трубочке [40]. Когда усиливается ток мочи, секреция АДГ уменьшается, и проводимость Kir1.1 снижается за счет взаимодействия с CFTR через белок NHERF (Na+/H+ exchanger regulatory factor — регуляторный фактор обмена Na+/H+). Таким образом, секреция калия и его потери с мочой сокращаются.
Эпителий дистального извитого канальца играет важную роль в реабсорбции Na+ [38]. В этом процессе участвуют натриевые каналы на апикальной мембране, работающие за счет концентрационного градиента, создаваемого Na+/K+-АТФазой на базолатеральной мембране. Гетеромеры Kir4.1/5.1 колокализуются с Na+/K+-АТФазой [22, 23, 41] и осуществляют рециклизацию K+, способствуя тем самым непрерывному транспорту Na+. У мыши эти каналы ингибируются закислением внутриклеточной среды с pKa ~7.6 [23]. Kir4.1 также служит посредником эффектов потребления натрия и калия на экспрессию и активность NCC (Na-Cl cotransporter). При низком потреблении натрия ток через Kir4.1/5.1 высокий, мембрана эпителия при этом гиперполяризована, общий уровень NCC и его фосфорилированной формы высокий, и Na+ реабсорбируется. При высоком потреблении Na+ ток внутреннего выпрямления и экспрессия NCC уменьшается. Мыши, в почках которых отсутствует Kir4.1, не способны регулировать активность NCC в зависимости от содержания Na+ в пище [42] и поэтому страдают от гипокалиемии и гипертензии.
В апикальной мембране основных клеток собирательной трубочки Kir1.1 обеспечивает секрецию K+ в мочу [36]. В базолатеральной мембране этих клеток присутствуют гетеромеры Kir4.1/5.1, которые, по-видимому, так же, как и в дистальном извитом канальце, поддерживают работу Na+/K+-АТФазы [43].
Kir4.1 в желудке
H+/K+-АТФаза в составе тубуловезикул встраивается в апикальную мембрану париетальных клеток желудка при их стимуляции гистамином, гастрином или ацетилхолином и обменивает K+ на H+. K+ поступает в просвет протока через калиевые каналы, которые также активируются при стимуляции клеток. Источником H+ служит CO2, который карбоангидраза превращает в H2CO3, а далее H2CO3 диссоциирует на H+ и HCO3−. HCO3− выходит из клетки через HCO3−/Cl− обменники на базолатеральной мембране. NKCC1 — гомолог почечного NKCC2 — также находящийся на базолатеральной мембране, обеспечивает приток в клетку Cl−, который затем выходит из клетки через хлорные каналы на апикальной мембране. Кроме того, на базолатеральной мембране расположена Na+/K+-АТФаза, ассоциированная с K+ каналами, которая в конечном счете поддерживает концентрационные градиенты [44].
Kir4.1 расположены на апикальной мембране париетальных клеток желудка, где колокализуются с H+/K+-АТФазой, и в ранних работах высказывалось предположение, что Kir4.1 действует синергично с потенциалзависимыми калиевыми каналами KCNQ1/KCNE2, поставляя K+ к внеклеточному сайту АТФазы [45, 46]. Однако у нокаутных по Kir4.1 животных слизистая оболочка желудка секретировала больше кислоты при более высокой экспрессии H+/K+-АТФазы на поверхности клеток [47]. Возможной причиной этих неожиданных результатов может быть дефект эндоцитоза, так как у нокаутных мышей тубуловезикулы не обнаруживаются, и H+/K+-АТФаза перманентно находится на апикальной мембране. В норме же Kir4.1 является хорошим кандидатом на калиевую проводимость, способствующую секреции кислоты, поскольку он практически не чувствителен к колебаниям pH [44, 45].
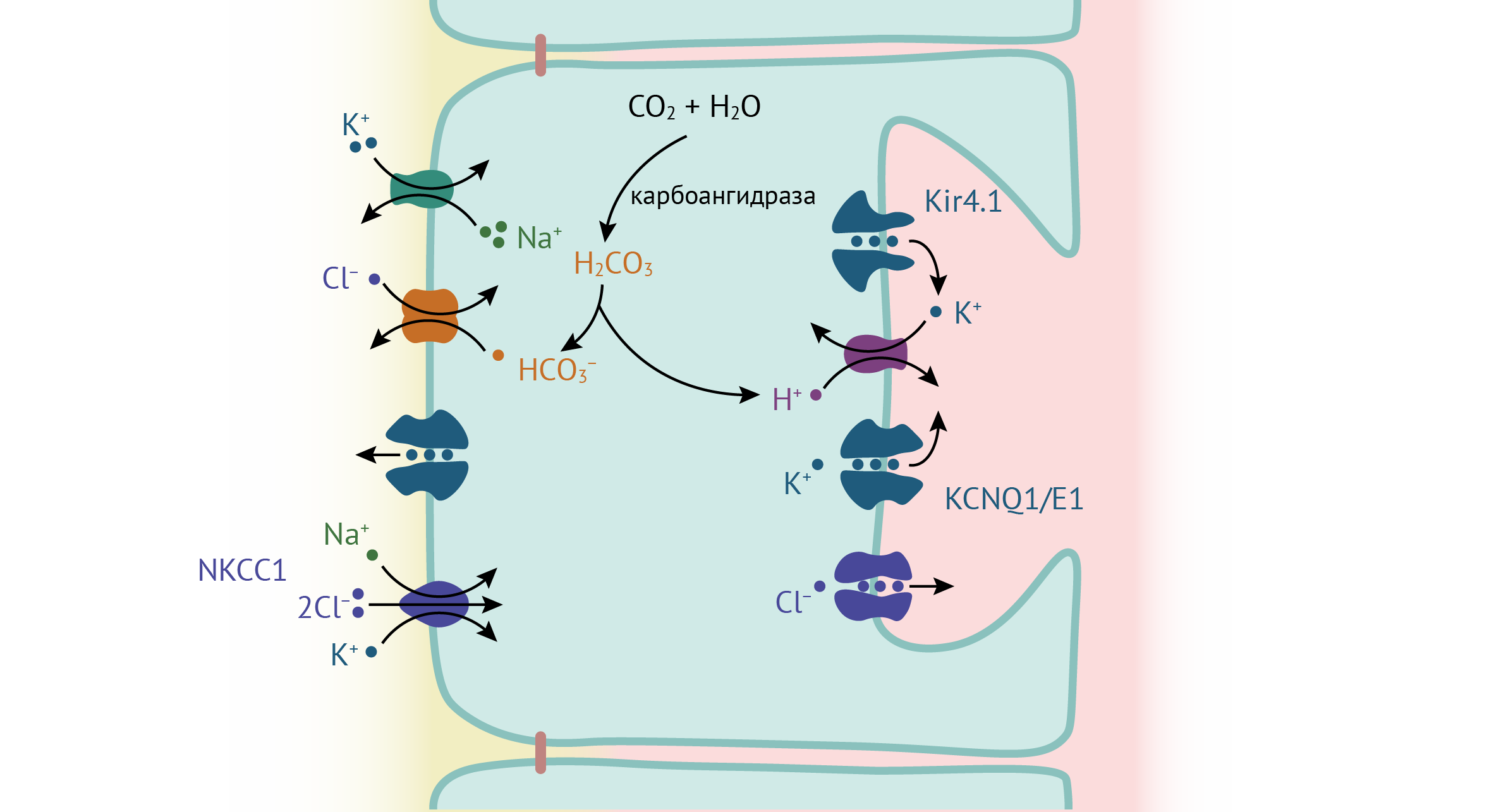
Рисунок 13. Роль Kir4.1 в секреции кислоты париетальными клетками эпителия желудка [44].
Kir4.1 и Kir5.1 во внутреннем ухе
Во внутреннем ухе лестница преддверия и барабанная лестница заполнены перилимфой, мало отличающейся по составу от обычной тканевой жидкости, однако эндолимфа спирального канала отличается очень высокой концентрацией K+, достигающей ~150 мМ, а значит, высоким электрическим потенциалом (~ +80 мВ) относительно крови или перилимфы; этот потенциал называют эндокохлеарным потенциалом (см. рис. 14) [48, 49]. Ионный состав и электрический потенциал эндолимфы необходимы для нормального слуха. K+ поступает в эндолимфу через латеральную стенку улитки, которая состоит из сосудистой полоски и спиральной связки. Kir4.1 экспрессируется в эпителии сосудистой полоски [50] на апикальной мембране промежуточных клеток [51, 52]. Нокаут Kcnj10−/− у мышей приводит к глухоте, сопровождающейся снижением концентрации K+ в эндолимфе вдвое и нулевым эндокохлеарным потенциалом [53]. На основании этих данных сформировалось представление, что Kir4.1 создает высокий диффузионный потенциал на апикальной мембране промежуточных клеток и формирует эндокохлеарный потенциал [48, 54, 55].
Kir5.1 экспрессируется в фиброцитах спиральной связки и локализован в основном внутриклеточно [51], однако перфузия внешних каналов раствором с Ba2+ несколько повышает эндокохлеарный потенциал. Из этого наблюдения родилось предположение, что часть Kir5.1 достигает плазматической мембраны и служит отрицательным регулятором эндокохлеарного потенциала [51, 56].
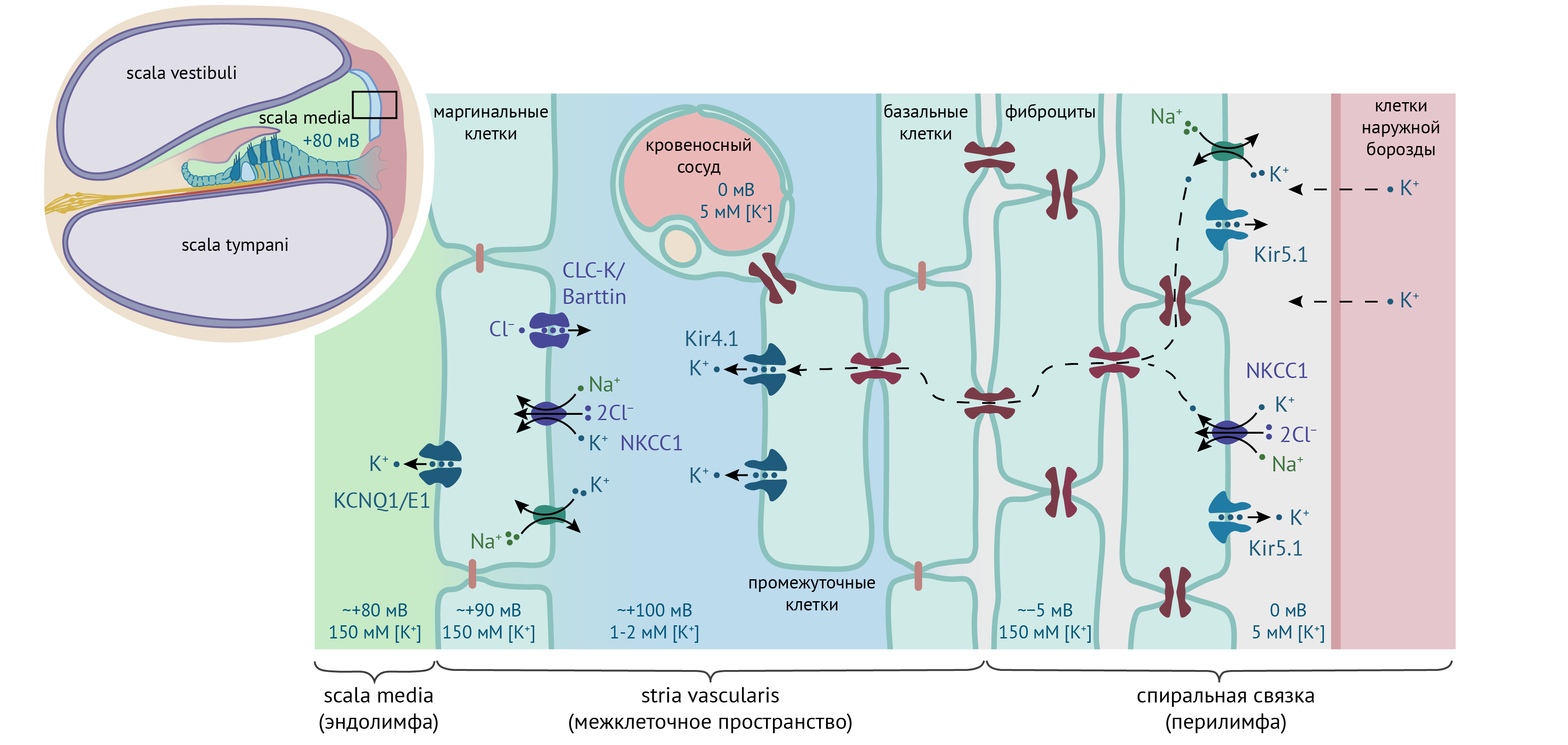
Рисунок 14. Роль Kir4.1 и Kir5.1 в поддержании эндокохлеарного потенциала (пояснения в тексте). Темно-красным показаны щелевые контакты. По [48].
Kir4.1 и Kir5.1 в нервной системе
Астроциты и клетки Мюллера
Важнейшей функцией астроглии и клеток Мюллера является поддержание ионного и осмотического гомеостаза внеклеточной среды. Они переносят K+ из участков с высокой концентрацией (активно работающих синапсов) в области с низким [K+]o [20]. Без механизма оттока K+ активно работающие синапсы будут постоянно деполяризованы и не смогут нормально функционировать. Для удаления K+ из перисинаптической области астроциты экспрессируют различные Kir каналы в различных участках мембраны. Основным участником этого процесса является Kir4.1 сам по себе либо вместе с Kir5.1: в гиппокампе нокаутных по Kir4.1 мышей нарушено удаление K+ астроцитами из области синапсов в ответ на стимуляцию нейронов [57].
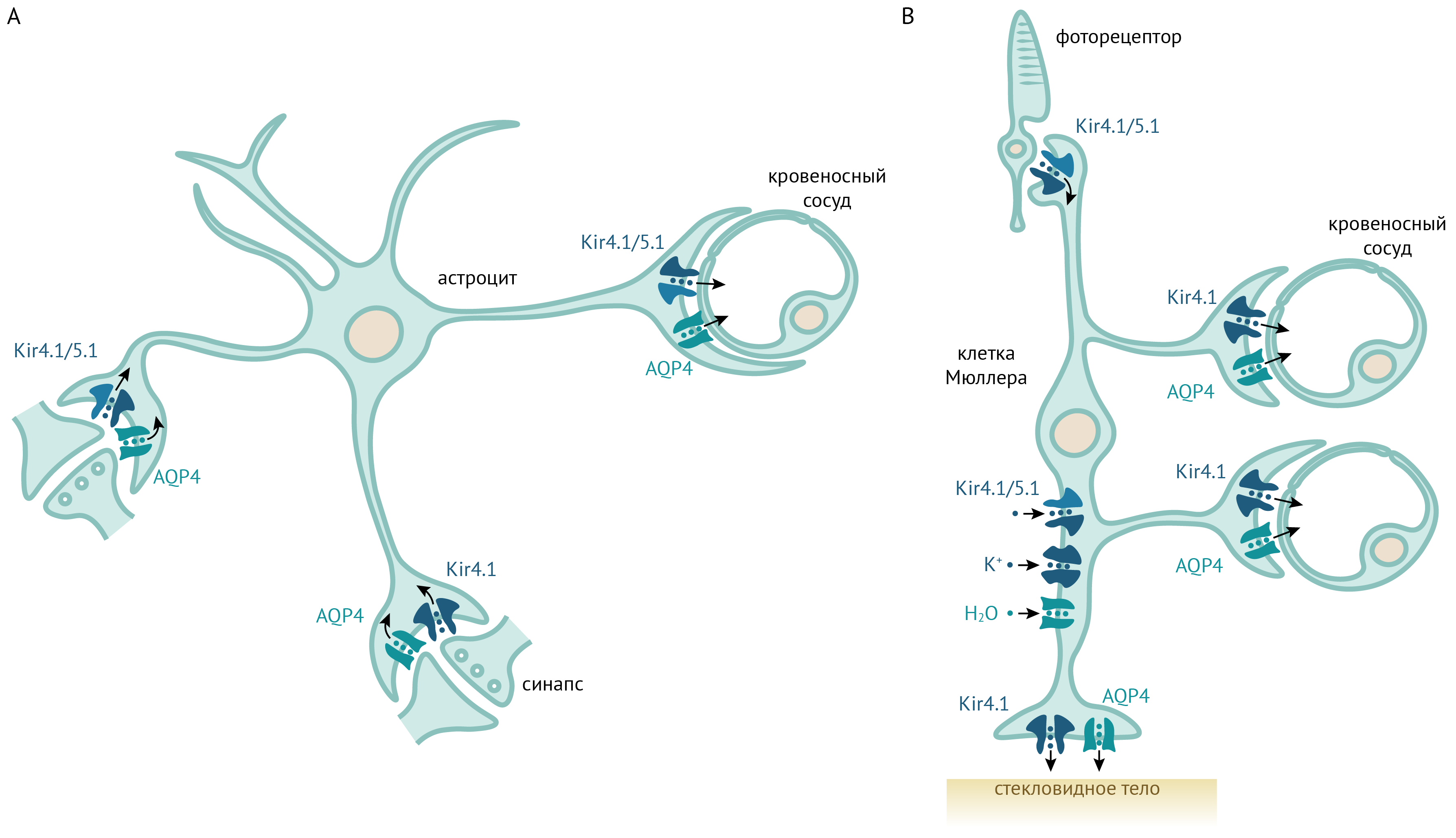
Рисунок 15. Роль Kir4.1 и Kir5.1 в астроцитах и клетках Мюллера. По [19]. K+, выделяемый активно работающими синапсами, поступает в астроциты через Kir4.1 и Kir4.1/5.1 и выводится через Kir4.1/5.1 через пластинчатые окончания отростков астроцитов, прилегающие к кровеносным сосудам. В сетчатке K+ выделяется фоторецепторами и через клетки Мюллера выходит в кровеносные сосуды и стекловидное тело.
В ЦНС и в сетчатке Kir4.1 и Kir5.1 экспрессируются как в виде гомомеров, так и в виде гетеромеров и распределены между различными участками плазматической мембраны.
В коре головного мозга Kir4.1/5.1 и Kir4.1 находятся на отростках астроцитов, окружающих синапсы. Однако на пластинчатых окончаниях отростков астроцитов («ножках» астроцитов), подходящих к мягкой оболочке мозга и кровеносным сосудам, присутствуют только гетеромеры. То есть K+ заходит в клетку и через гомомеры, и через гетеромеры, а покидает астроцит через гетеромерные каналы (см рис. 15).
Основное функциональное различие между гомо- и гетеромерными каналами заключается в чувствительности последних к внутриклеточному pH. В астроцитах с высокой экспрессией Na+/HCO3− котранспортера его активность приводит к гиперполяризации мембраны и повышению внутриклеточного pH (на один ион Na+ он переносит два-три HCO3−). Деполяризация мембраны астроцита при активной работе синапса стимулирует котранспортер, внутриклеточный pH повышается и стимулирует ток через Kir4.1/5.1, усиливая поступление K+ в астроцит. В тех отделах мозга, где экспрессия Na+/HCO3− котранспортера низкая (например, в таламусе и гиппокампе), преобладают гомомеры Kir4.1 [58].
Появляются свидетельства роли астроцитарного Kir4.1 в патогенезе хореи Хантингтона [59] и депрессии [60]. В стриатуме мышей, служащих моделью хореи Хантингтона, экспрессия Kir4.1 в астроцитах была снижена. Низкий уровень IK в астроцитах сопровождался двукратным повышением [K+]o. Инъекция гена Kcnj10 в составе аденовирусного вектора в стриатум этих мышей нормализовала электрофизиологические показатели астроцитов и внеклеточную концентрацию K+, улучшало моторные функции и выживаемость животных [59].
Исследовали сравнили экспрессию различных генов в латеральных ядрах поводков (LHb) у нормальных крыс и в модели депрессии и обнаружили значительное повышение экспрессии Kir4.1. Само по себе повышение экспрессии Kir4.1 при инъекции гена Kcnj10 под астроцитарным промотором в составе аденовируса, избирательно заражающего астроциты, вызвало у крыс симптомы депрессии. В норме для значительной доли нейронов LHb характерна тоническая активность, однако при повышении уровня Kir4.1 в тесно окружающих эти нейроны астроцитах удаление K+ из ограниченного пространства между нейронами и астроцитами усиливается. Снижение [K+]o, согласно уравнению Нернста, приводит к гиперполяризации нейронов и изменению характера их активности: вместо регулярных одиночных ПД возникают более редкие всплески активности. Исследователям удалось снять симптомы депрессии путем снижения экспрессии Kir4.1 в астроцитах с помощью инъекции аденовирусного вектора, содержащего малую РНК, образующую шпильки, (shRNA) против транскрипта Kcnj10 или доставки в астроциты LHb доминантно-негативного варианта Kir4.1. В предложенной авторами модели депрессии кетамин, блокируя NMDAR (а значит, деполяризующие токи), не позволяет возникать всплескам активности в нейронах LHb и возвращает их к тонической активности, тем самым облегчая симптомы депрессии [60].
В сетчатке клетки Мюллера экспрессируют гомомер Kir4.1 на отростках, обращенных к стекловидному телу и кровеносным сосудам, но только гетеромеры на перисинатических отростках [61]. Такое распределение подсказывает, что калий входит в клетки Мюллера через гетеромеры, а выходит из них через гомомеры Kir4.1.
На секретирующей K+ стороне глиальных клеток Kir каналы функционально связаны с аквапоринами AQP4, нокаут которых замедляет ток K+ через астроциты [62].
Олигодендроциты
Помимо астроглии, Kir4.1 экспрессируется в олигодендроцитах, причем как в их телах, так и в миелине, на стороне, обращенной к аксону [63]. Для изучения роли Kir4.1 в олигодендроцитах авторы исследования использовали две олигодендроцит-специфичные Cre-линии (то есть линии мышей, в олигодендроцитах которых экспрессируется Cre-рекомбиназа, которая может вырезать гены, окруженные loxP сайтами): Olig2-Cre, в которой рекомбинация происходит в более раннем возрасте, и Cnp-Cre, которая активна в более позднем возрасте. С помощью первой линии они выяснили, что Kir4.1 не требуется для нормальной миелинизации аксонов в раннем возрасте. Используя обе линии, они показали, что Kir4.1 не обязателен для ремиелинизации волокон после острой демиелинизации, однако необходим для поддержания целостности миелиновых оболочек. Нокаутные животные показывают худшие результаты в тестах на двигательные функции, а удлиненное латентное время вызванных зрительных потенциалов свидетельствует о нарушениях зрения [63].
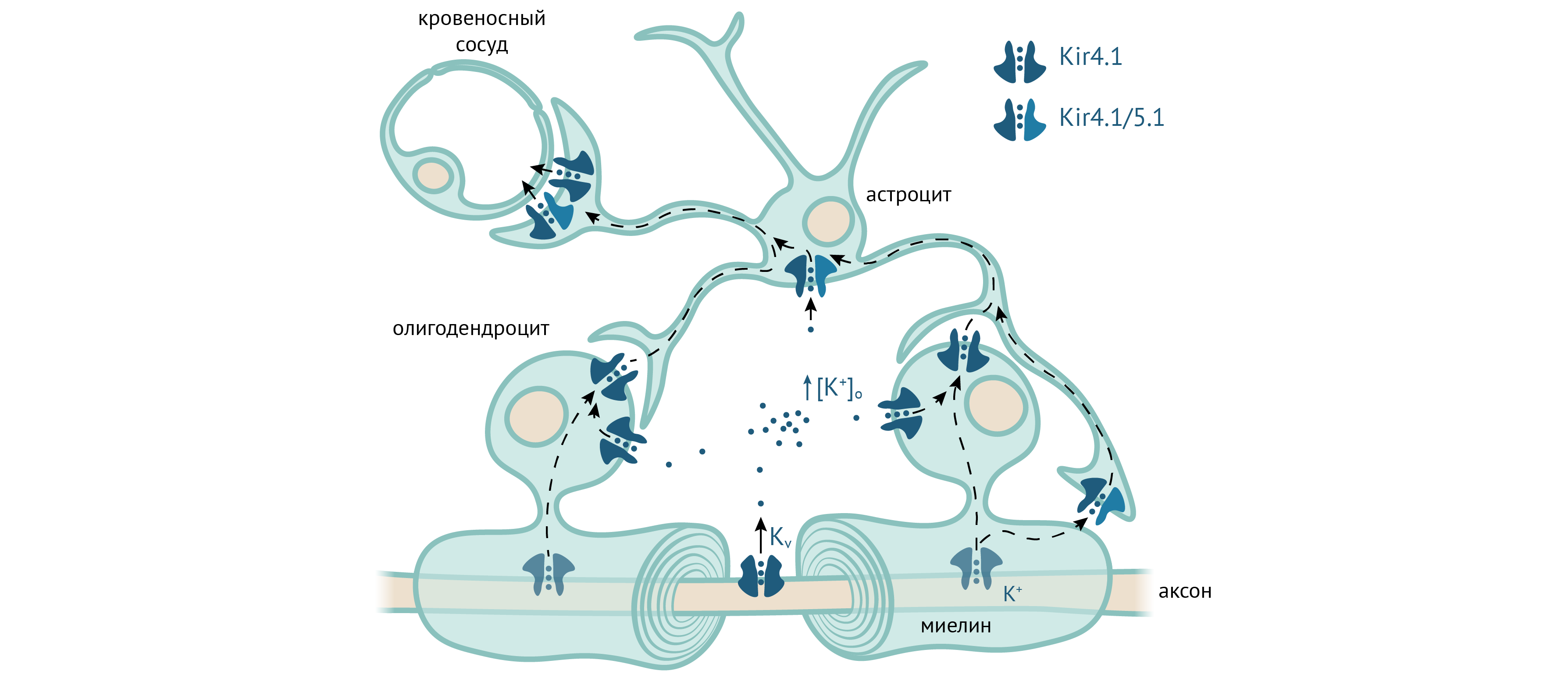
Рисунок 16. Роль Kir4.1 и Kir5.1 в олигодендроцитах. Олигодендроциты поглощают K+, поступающий из аксона, и выделяют его для последующего удаления астроцитами. По [63].
Нейроны голубого пятна
Голубое пятно, locus coeruleus, содержит норадренергические нейроны и участвует в процессах внимания, обучения, памяти и ответах на стресс и боль [64, 65]. Кроме того, нейроны этого ядра стимулируют дыхание в ответ на стимуляцию CO2 [66]. В норме основной функцией pH-чувствительных нейронов голубого пятна, вероятно, является запуск тревоги или аверсии в ответ на гиперкапнию.
Гетеромеры Kir4.1/5.1 экспрессируются в хемочувствительных ядрах мозга, в том числе в нейронах голубого пятна. Kcnj16−/− мыши, в отличие от Kcnj10−/−, доживают до взрослого возраста и не имеют очевидных нарушений физиологии и поведения. Нейроны голубого пятна этих мышей на ~75 % слабее реагируют на закисление внутриклеточной среды [67]. В голубом пятне экспрессируются и другие каналы, чувствительные к pH: TASK-1 [68, 69] чувствителен к изменению внеклеточного pH и может опосредовать остаточный ответ в нокаутных нейронах. Однако гипотеза о роли Kir4.1/5.1 в ответе на гиперкапнию в отсутствие поведенческих данных требует дальнейшей проверки.
Kir7.1 в различных тканях
Kir7.1 экспрессируется в различных эпителиях: в сосудистом сплетении [28, 70, 71] и в пигментном эпителии сетчатки [72, 73], в фолликулярных клетках щитовидной железы, в эпителии почечных канальцев (в проксимальном канальце, дистальном извитом канальце и в собирательной трубочке), в эпителии носовой полости и дыхательных путей, в эпителии подвздошной кишки {1} [74]. Kir7.1 всегда колокализуется с Na+/K+-АТФазой. В эпителиях с «обратной» полярностью — в сосудистом сплетении и пигментном эпителии сетчатки — они расположены на апикальной мембране, а в эпителиях с «нормальной» полярностью — на базолатеральной {2} [70, 74]. Колокализация с Na+/K+-АТФазой послужила основанием для гипотезы о том, что Kir7.1 участвует в рециклизации калия, который захватывается АТФазой для предотвращения накопления K+ и поддержания трансэпителиального транспорта — роль, подобная другим транспортным каналам Kir [44].
Попытка получить мышей, нокаутных по Kcnj13−/−, выявила участие Kir7.1 в развитии носовой полости и дыхательных путей. Kcnj13−/− мыши погибали сразу после рождения из-за несращения твердого неба и, вероятно, из-за трудностей с дыханием вследствие задержки развития легких [75]. Однако эти результаты надо с осторожностью переносить на человека, так как у человека гомозиготные нонсенс-мутации Arg166X и Trp53X не летальны и не сопровождаются несращением твердого неба [76, 77].
В другом исследовании [78] были отобраны случайные мутации, приводящие к нарушению развития трахеи у мышей, секвенирование мутантных животных показало, что у них была мутация Kir7.1Leu13Pro. У мутантных мышей трахея была короче, а также было нарушено развитие хрящевых колец. Кроме того, у этих мышей был укорочен пищевод, который также является производным передней кишки. Детальное исследование этого фенотипа показало, что недоразвитие трахеи и пищевода вызвано неправильным упорядочением гладкомышечных клеток вследствие снижения фосфорилирования AKT и вызванного этим нарушения выстраивания актинового цитоскелета.
Мыши с мозаичной экспрессией Kcnj13, полученные методом CRISPR-Cas9 в поколении F0 [79], а затем и мыши с тканеспецифичным нокаутом Kcnj13 в пигментном эпителии [80] послужили хорошей системой для оценки роли Kir7.1 в пигментном эпителии сетчатки. Фоторецепторы, находящиеся напротив клеток пигментного эпителия, не окрашенных антителом против Kir7.1, дегенерируют, и иммунореактивность к родопсину появляется во внутренних сегментах фоторецепторов [79]. Вероятно, выход K+ через Kir7.1 участвует в поддержании ионного гомеостаза внешних сегментов фоторецепторов. В темноте открыты цГМФ-управляемые катионные каналы во внешних сегментах фоторецепторов, которые пропускают Na+ и Ca2+, и их вход в клетку компенсируется выходом K+ во внутренних сегментах фоторецепторов. На свету цГМФ-управляемые катионные каналы закрываются, и выход K+ замедляется. Кроме того, Na+/K+-АТФаза, расположенная на мембране внутренних сегментов, закачивает K+ в клетку, и концентрация K+ в пространстве между пигментным эпителием и фоторецепторами снижается с 5 мМ до 2 мМ. Это падение концентрации сдвигает равновесный потенциал для K+ к более отрицательным значениям и приводит к усилению калиевого тока и торможению работы Na+/K+-АТФазы на апикальной мембране клеток пигментного эпителия. Таким образом, внеклеточная концентрация K+ возвращается к нормальным значениям [81].
Еще один эпителий с высокой экспрессией Kir7.1 — это сосудистые сплетения в желудочках мозга. Предполагается, что в сосудистом сплетении Kir7.1 (и Kv1) участвует в секреции цереброспинальной жидкости и поддержании в ней низкой концентрации K+ [82]. Na+/K+-АТФаза закачивает K+ в клетки сосудистого сплетения и создает градиент для работы NKCC1 на апикальной мембране, который выводит K+ из цереброспинальной жидкости. Kir7.1 поставляет калий к АТФазе и к NKCC1.
У Danio rerio мутация Kir7.1 вызывает вариант окраски jaguar/obelix. Меланосомы рыб с этой мутацией неспособны адекватно отвечать на сигналы рассредоточения меланосом — помещение на темный фон после светлого (см. рис. 17) или применение антагониста a2-адренорецепторов [83].
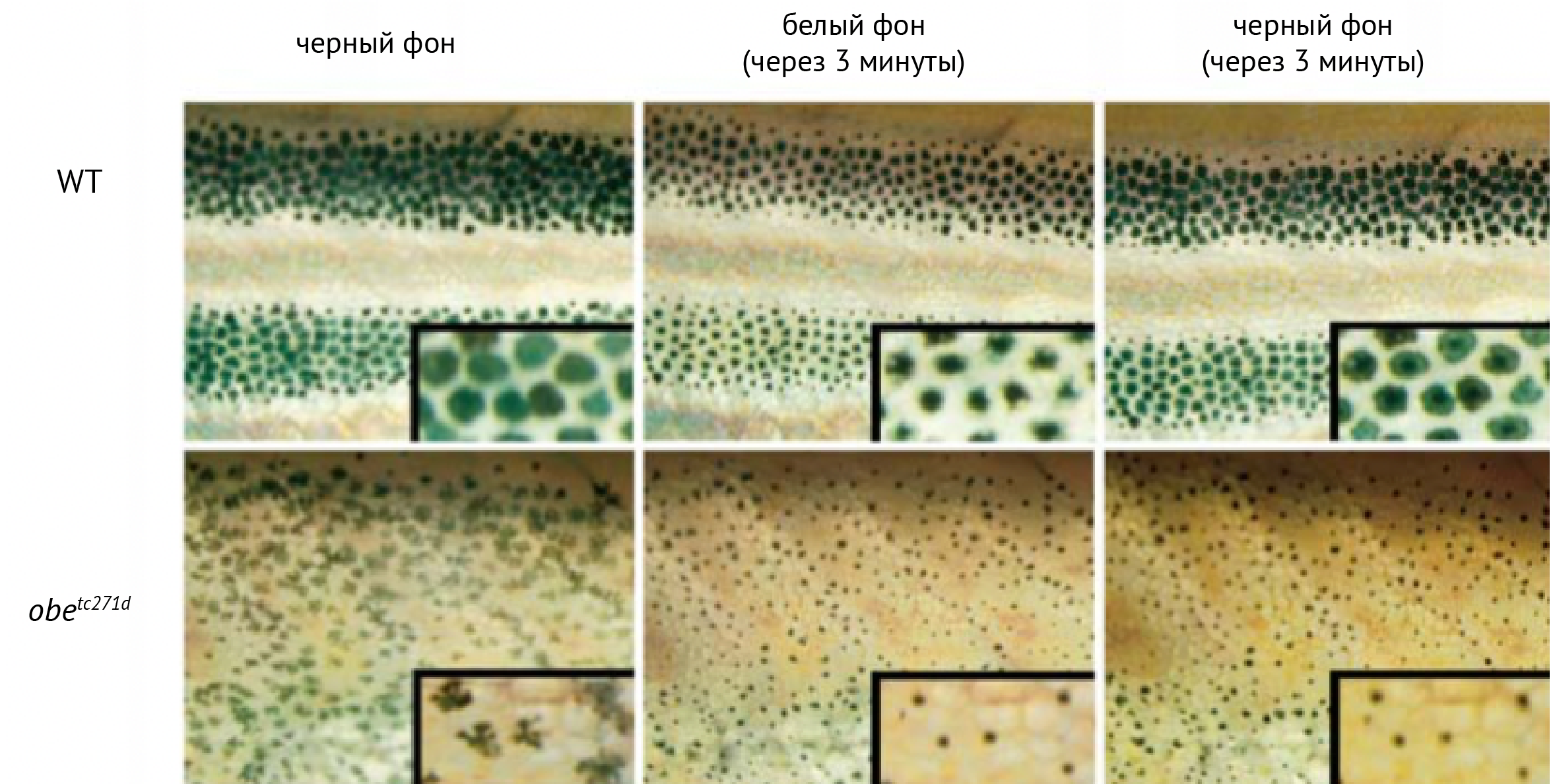
Рисунок 17. Меланосомы рыб дикого типа (WT) собираются вместе при помещении рыбы на светлый фон, повторное помещение на темный фон вызывает рассредоточение меланосом. У мутантных рыб меланосомы не рассредотачиваются при помещении на темный фон после светлого [83].
Кроме эпителиальных клеток, функциональное значение Kir7.1 было показано для миометрия матки: в середине беременности высокий уровень экспрессии Kir7.1 поддерживает мембранный потенциал миоцитов близким к EK, но на более поздних сроках экспрессия Kir7.1 падает, и мембрана деполяризуется до −45 мВ, стимулируя сокращения. Также в этой работе было показано, что применение блокатора Kir7.1 VU590 на мышах и на полосках человеческого миометрия вызывает более продолжительные сокращения миометрия, чем применение окситоцина [84].
Мутации в транспортных каналах Kir
Kir1.1 — синдром Барттера
Синдром Барттера — это патология почечных канальцев, симптомами которой являются гипокалиемия, метаболический алкалоз, излишнее выделение солей почками, повышение уровня ренина и альдостерона [85, 86]. К этому фенотипу могут приводить мутации в различных каналах и транспортерах, на основании чего синдром Барттера делится на четыре типа. При мутациях в Kir1.1 говорят о синдроме Барттера типа II (OMIM: 241200) [38, 87, 88].
Алкалоз и повышение уровня ренина и альдостерона вызваны нарушением реабсорбции NaCl. Для компенсации дефекта реабсорбции Na+ почка выделяет ренин, превращающий ангиотензиноген в ангиотензин I. Ангиотензин I под действием ангиотензин-превращающего фермента переходит в ангиотензин II, который действует на надпочечники, стимулируя секрецию альдостерона. Альдостерон в свою очередь усиливает реабсорбцию Na+ в дистальном нефроне через ENaC, при этом увеличивается электродвижущая сила для секреции K+ и H+, что ведет к гипокалиемии и алкалозу. Почему при синдроме Барттера возникает гипокалиемия, если Kir1.1 опосредует секрецию калия? У младенцев с этим синдромом иногда наблюдается преходящая гиперкалиемия [89, 90]. Ответ на этот вопрос был дан при изучении нокаутных мышей: нокаут Kir1.1 нарушает реабсорбцию K+ в отделе восходящего колена петли Генле через NKCC2, однако секреция через BK (big conductance K+ channel — калиевый канал с высокой проводимостью) и Ca2+-активируемые K+ каналы в дистальном извитом канальце сохраняется.
Мутации в Kir4.1 — EAST/SeSAME синдром
Мутации KCNJ10 у человека вызывают EAST (epilepsy, ataxia, sensorineural deafness, tubulopathy) синдром, также называемый SeSAME (seizures, sensorineural deafness, ataxia, mental retardation, electrolyte imbalances) синдромом, (OMIM: 612780) — заболевание, наследуемое по аутосомно-рецессивному типу и сопровождающееся судорогами, нейросенсорной глухотой, атаксией, задержкой умственного развития и дисбалансом электролитов (гипокалиемия, метаболический алкалоз и гипомагниемия) [91, 92]. Все эти симптомы хорошо укладываются в представления о роли Kir4.1 в различных тканях. Судороги возникают из-за неспособности астроцитов без Kir4.1 к отведению K+ от области синапсов, при этом астроциты и нейроны в этих областях деполяризуются и порог возбуждения снижается. Кроме того, деполяризованные астроциты не способны к эффективному захвату глутамата, что тоже ведет к снижению судорожного порога. В более раннем исследовании было высказано предположение о связи полиморфизмов в KCNJ10 со склонностью к судорожным припадкам [93], однако реальное влияние этих полиморфизмов не выяснено.
Потеря слуха у пациентов, вероятно, связана со снижением эндокохлеарного потенциала, как это происходит и с нокаутными животными. Атаксия может возникать из-за нарушений в мозжечке (в пользу этого предположения говорит уменьшенный объем мозжечка у некоторых пациентов) или из-за потери проприоцепции, однако разделить эти возможности трудно вследствие когнитивных нарушений.
Биопсия нерва одного из пациентов показала гипомиелинизацию, что согласуется с утратой миелинизации у нокаутных животных.
Утрата Kir4.1 может вызывать потерю солей с мочой через торможение работы Na+/K+-АТФазы в дистальном извитом канальце. Сниженный трансэпителиальный потенциал затрудняет реабсорбцию Na+ и Cl−, а также Mg2+, которые выходят с мочой. Алкалоз и повышение уровня ренина и альдостерона происходят так же, как при синдроме Барттера [91]. Мутации, обнаруженные у пациентов, нарушают функцию канала различными способами как в гомомерном состоянии, так и в составе гетеромеров Kir4.1/5.1 [94, 95].
Есть сообщения о дигенных мутациях KCNJ10 совместно с другими транспортными белками, которые вызывают различные фенотипы в гетерозиготном состоянии. Так, Yang и соавторы [96] обнаружили двух пациентов с несиндромальной тугоухостью с мутациями в KCNJ10 и SLC26A4 — известном гене, мутации в котором (в гомозиготном состоянии) ассоциированы с этим состоянием. В случае, описанном Hasan et al. [97], пациент с двумя миссенс-мутациями в генах KCNJ10 и KCNT1 в гетерозиготном состоянии страдал от тяжелых судорожных припадков и задержки развития.
Мутации в Kir7.1 — заболевания сетчатки
Мутации KCNJ13 у человека связаны с различными заболеваниями сетчатки.
Инеевидная дегенерация сетчатки наследуется по аутосомно-доминантному типу и характеризуется нитевидной дегенерацией стекловидного тела, ранней катарактой, мелкими кристаллическими отложениями в сетчатке и отслоением сетчатки. Мутантный Kir7.1Arg162Trp у пациентов с этой патологией опосредует неселективный катионный ток, который деполяризует мембраны клеток пигментного эпителия сетчатки [98]. Мутантные субъединицы не образуют функциональных гомотетрамеров, однако могут встраиваться гетеротетрамеры с субъединицами дикого типа и изменять их свойства в первую очередь через изменение конформации субъединицы вследствие нарушения взаимодействия с PIP2 [99].
Другие мутации утраты функции в KCNJ13 вызывают амавроз Лебера (LCA16) — утрату фоторецепторов в раннем детстве. C этим заболеванием связаны миссенс-мутация Leu241Pro и нонсенс-мутации Arg166X и Trp53X [76, 77], также были обнаружены и другие мутации у пациентов с пигментным ретинитом с более поздним развитием [76].
В Саудовской Аравии описана серия случаев витреоретинальной дистрофии и ранней катаракты. Пациенты в этом исследовании оказались носителями одной и той же рецессивной мутации Kir7.1Ile120Thr в гомозиготном состоянии, в то время как гетерозиготные родители двух гомозиготных дочерей были здоровы [100]. Поскольку мутация проявляется только в гомозиготном состоянии, можно предположить, что это мутация утраты функции, однако функционального исследования этой мутации проведено не было.
Дополнения
{1} Какими методами определяют локализацию канала? Некоторые выводы об экспрессии белка можно сделать по количеству его транскриптов, измеренных с помощью количественной ПЦР с обратной транскрипцией (qRT-PCR) или гибридизации in situ, однако количество транскриптов имеет опосредованное отношение к экспрессии белка. Также этот метод не позволяет определить субклеточную локализацию белка, поэтому такие исследования сопровождались электрофизиологическими измерениями с изолированных участков мембраны и сравнением характеристик тока с током в гетерологичной системе экспрессии. Для выяснения локализации белка напрямую можно получить антитела к белку, однако до настоящего времени получение антител к интегральным белкам ионных каналов, которые работали бы не только для вестерн-блота, но и для иммуногистохимического окрашивания, остается затруднительным. С разработкой методики CRISPR-Cas9 для получения трансгенных мышей все чаще получают мышей, у которых изучаемый белок помечен тегом (небольшим аминокислотным фрагментом). Тег может служить эпитопом для стандартизованных антител (например, HA или Myc-теги), а может быть флуоресцентным (например, GFP), однако при низких уровнях экспрессии для детекции используют антитела к GFP.
{2} Полярность эпителия определяется по набору транспортных белков, характерных для апикальной или базолатеральной мембраны, в первую очередь, по Na+/K+-АТФазе, которая в нормальных эпителиях располагается на базолатеральной мембране.
Библиография
1. Boim M.A. et al. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms // Am. J. Physiol. Physiol. 1995. Vol. 268, № 6 Pt 2. P. F1132-40.
2. Chepilko S. et al. Permeation and gating properties of a cloned renal K+ channel // Am. J. Physiol. 1995. Vol. 268, № 2 Pt 1. P. C389-401.
3. Choe H. et al. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. // Am. J. Physiol. 1997. Vol. 273, № 4 Pt 2. P. F516-29.
4. Doi T. et al. Extracellular K+ and Intracellular pH Allosterically Regulate Renal Kir1.1 Channels // J. Biol. Chem. 1996. Vol. 271, № 29. P. 17261–17266.
5. Fakler B. et al. Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. // EMBO J. 1996. Vol. 15, № 16. P. 4093–4099.
6. McNicholas C.M. et al. pH-dependent modulation of the cloned renal K+ channel, ROMK // Am. J. Physiol. 1998. Vol. 275, № 6 Pt 2. P. F972-81.
7. Tsai T.D. et al. Intracellular H+ inhibits a cloned rat kidney outer medulla K+ channel expressed in Xenopus oocytes // Am. J. Physiol. 1995. Vol. 268, № 5 Pt 1. P. C1173-8.
8. Wang R. et al. Subunit stoichiometry of the Kir1.1 channel in proton-dependent gating // J. Biol. Chem. 2005. Vol. 280, № 14. P. 13433–13441.
9. Schulte U. et al. pH gating of ROMK (Kir1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome // Proc. Natl. Acad. Sci. 1999. Vol. 96, № 26. P. 15298–15303.
10. Peters M. et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome // Kidney Int. 2003. Vol. 64, № 3. P. 923–932.
11. Rapedius M. et al. Structural and functional analysis of the putative pH sensor in the Kir1.1 (ROMK) potassium channel // EMBO Rep. 2006. Vol. 7, № 6. P. 611–616.
12. Ma D. et al. Role of ER export signals in controlling surface potassium channel numbers // Science. 2001. Vol. 291, № 5502. P. 316–319.
13. Connell A.D.O. et al. Phosphorylation-regulated endoplasmic reticulum retention signal in the renal outer-medullary K+ channel (ROMK) // Proc. Natl. Acad. Sci. 2005. Vol. 102, № 28. P. 9954–9959.
14. Zeng W.-Z. et al. Evidence for endocytosis of ROMK potassium channel via clathrin-coated vesicles // Am. J. Physiol. Physiol. 2002. Vol. 283, № 4. P. F630–F639.
15. Takumi T. et al. A Novel ATP-dependent Inward Rectifier Potassium Channel Expressed Predominantly in Glial Cells // J. Biol. Chem. 1995. Vol. 270, № 27. P. 16339–16346.
16. Bredt D.S. et al. Cloning and expression of two brain-specific inwardly rectifying potassium channels. // Proc. Natl. Acad. Sci. 1995. Vol. 92, № 15. P. 6753–6757.
17. Shuck M.E. et al. Cloning and Characterization of Two K+ Inward Rectifier (Kir) 1.1 Potassium Channel Homologs from Human Kidney (Kir1.2 and Kir.3) // J. Biol. Chem. 1997. Vol. 272, № 1. P. 586–593.
18. Bond C.T. et al. Cloning and expression of a family of inward rectifier potassium channels // Receptors and Channels. 1994. Vol. 2, № 3. P. 183–191.
19. Hibino H. et al. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles // Physiol. Rev. 2010. Vol. 90, № 1. P. 291–366.
20. Newman E.E.A. Regional specialization of retinal glial cell membrane // Nature. 1984. Vol. 309. P. 155–157.
21. Pessia M. et al. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels // EMBO J. 1996. Vol. 15, № 12. P. 2980–2987.
22. Tanemoto M. et al. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1 // J. Physiol. 2000. Vol. 525, № 3. P. 587–592.
23. Lourdel S. et al. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: Similarities with Kir4-Kir5.1 heteromeric channels // J. Physiol. 2002. Vol. 538, № 2. P. 391–404.
24. Yang Z. et al. Biophysical and Molecular Mechanisms Underlying the Modulation of Heteromeric Kir4.1–Kir5.1 Channels by CO2 and pH // J. Gen. Physiol. 2000. Vol. 116, № September 2014. P. 33–45.
25. Gosset P. et al. A new inward rectifier potassium channel gene (KCNJ15) localized on chromosome 21 in the Down syndrome chromosome region 1 (DCR1) // Genomics. 1997. Vol. 44, № 2. P. 237–241.
26. Pearson W.L. et al. Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver // J. Physiol. 1999. Vol. 514, № 3. P. 639–653.
27. Pessia M. et al. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1 // J. Physiol. 2001. Vol. 532, № 2. P. 359–367.
28. Döring F. et al. The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties // J. Neurosci. 1998. Vol. 18, № 21. P. 8625–8636.
29. Krapivinsky G. et al. A novel inward rectifier K+ channel with unique pore properties // Neuron. 1998. Vol. 20, № 5. P. 995–1005.
30. Partiseti M. et al. Cloning and characterization of a novel human inwardly rectifying potassium channel predominantly expressed in small intestine // FEBS Lett. 1998. Vol. 434, № 1–2. P. 171–176.
31. Rohacs T. et al. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels // Proc. Natl. Acad. Sci. 2003. Vol. 100, № 2. P. 745–750.
32. Hughes B.A., Swaminathan A. Modulation of the Kir7.1 potassium channel by extracellular and intracellular pH // Am. J. Physiol. Physiol. 2008. Vol. 294, № 2. P. C423–C431.
33. Kohda Y. et al. Localization of the ROMK potassium channel to the apical membrane of distal nephron in rat kidney // Kidney Int. 1998. Vol. 54, № 4. P. 1214–1223.
34. Ortega B. et al. Stable, polarised, functional expression of Kir1.1b channel protein in Madin-Darby canine kidney cell line // J. Physiol. 2000. Vol. 528, № 1. P. 5–13.
35. Xu J.Z. et al. Localization of the ROMK protein on apical membranes of rat kidney nephron segments // Am. J. Physiol. 1997. Vol. 273. P. F739–F748.
36. Lu M. et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice // J. Biol. Chem. 2002. Vol. 277, № 40. P. 37881–37887.
37. Lorenz J.N. et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome // J. Biol. Chem. 2002. Vol. 277, № 40. P. 37871–37880.
38. Hebert S.C. et al. Molecular Diversity and Regulation of Renal Potassium Channels // Physiol. Rev. 2005. Vol. 85, № 1. P. 319–371.
39. Lu M. et al. CFTR is required for PKA-regulated ATP sensitivity of Kir1.1 potassium channels in mouse kidney // J. Clin. Invest. 2006. Vol. 116, № 3. P. 797–807.
40. Field M.J., Stanton B.A., Giebisch G.H. Influence of ADH on renal potassium handling: A micropuncture and microperfusion study // Kidney Int. 1984. Vol. 25, № 3. P. 502–511.
41. Tucker S.J. et al. pH Dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia // J. Biol. Chem. 2000. Vol. 275, № 22. P. 16404–16407.
42. Wu P. et al. Kir4.1/Kir5.1 Activity Is Essential for Dietary Sodium Intake–Induced Modulation of Na-Cl Cotransporter // J. Am. Soc. Nephrol. 2018. Vol. 30, № 2. P. 216–227.
43. Lachheb S. et al. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells // Am. J. Physiol. Physiol. 2008. Vol. 294, № 6. P. F1398–F1407.
44. Sepúlveda F. V. et al. Molecular Aspects of Structure, Gating, and Physiology of pH-Sensitive Background K2P and Kir K+ -Transport Channels // Physiol. Rev. 2015. Vol. 95, № 1. P. 179–217.
45. Fujita A. et al. Specific localization of an inwardly rectifying K+ channel, Kir4.1, at the apical membrane of rat gastric parietal cells; its possible involvement in K+ recycling for the H+-K+-pump // J. Physiol. 2002. Vol. 540, № 1. P. 85–92.
46. Kaufhold M.A. et al. Localization, Trafficking, and Significance for Acid Secretion of Parietal Cell Kir4.1 and KCNQ1 K+ Channels // Gastroenterology. 2008. Vol. 134, № 4. P. 1058–1069.
47. Song P. et al. Kir4.1 Channel Expression Is Essential for Parietal Cell Control of Acid Secretion // J. Biol. Chem. 2011. Vol. 286, № 16. P. 14120–14128.
48. Hibino H., Kurachi Y. Molecular and Physiological Bases of the K+ Circulation in the Mammalian Inner Ear // Physiology. 2006. Vol. 21, № 5. P. 336–345.
49. Békésy G. V. Resting potentials inside the cochlear partition of the guinea pig // Nature. 1952. Vol. 169, № 4293. P. 241–242.
50. Hibino H. et al. An ATP-dependent inwardly rectifying potassium channel, KAB-2 (Kir4.1), in cochlear stria vascularis of inner ear: its specific subcellular localization and correlation with the formation of endocochlear potential. // J. Neurosci. 1997. Vol. 17, № 12. P. 4711–4721.
51. Hibino H. et al. Expression of an inwardly rectifying K+ channel, Kir5.1, in specific types of fibrocytes in the cochlear lateral wall suggests its functional importance in the establishment of endocochlear potential // Eur. J. Neurosci. 2004. Vol. 19, № 1. P. 76–84.
52. Ando M., Takeuchi S. Immunological identification of an inward rectifier K+ channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis of gerbils and rats // Cell Tissue Res. 1999. Vol. 298, № 1. P. 179–183.
53. Marcus D.C. et al. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential // Am. J. Physiol. Physiol. 2013. Vol. 282, № 2. P. C403–C407.
54. Nin F. et al. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear // Proc. Natl. Acad. Sci. 2008. Vol. 105, № 5. P. 1751–1756.
55. Takeuchi S., Ando M., Kakigi A. Mechanism generating endocochlear potential: Role played by intermediate cells in stria vascularis // Biophys. J. 2000. Vol. 79, № 5. P. 2572–2582.
56. Marcus D.C. Characterization of potassium permeability of cochlear duct by perilymphatic perfusion of barium // Am. J. Physiol. Physiol. 1984. Vol. 247, № 3. P. C240–C246.
57. Djukic B. et al. Conditional Knock-Out of Kir4.1 Leads to Glial Membrane Depolarization, Inhibition of Potassium and Glutamate Uptake, and Enhanced Short-Term Synaptic Potentiation // J. Neurosci. 2007. Vol. 27, № 42. P. 11354–11365.
58. Schmitt B.M. et al. Na/HCO3 Cotransporters in Rat Brain: Expression in Glia, Neurons, and Choroid Plexus // J. Neurosci. 2000. Vol. 20, № 18. P. 6839–6848.
59. Tong X. et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice // Nat. Neurosci. 2014. Vol. 17, № 5. P. 694–703.
60. Cui Y. et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression // Nature. Nature Publishing Group, 2018. Vol. 554, № 7692. P. 323–327.
61. Ishii M. et al. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina // Am. J. Physiol. Physiol. 2003. Vol. 285, № 2. P. C260–C267.
62. Padmawar P. et al. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator // Nat. Methods. 2005. Vol. 2, № 11. P. 825–827.
63. Schirmer L. et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity // Elife. 2018. Vol. 7. P. 1–21.
64. Llorca-Torralba M. et al. Noradrenergic Locus Coeruleus pathways in pain modulation // Neuroscience. IBRO, 2016. Vol. 338. P. 93–113.
65. Benarroch E.E. Locus coeruleus // Cell Tissue Res. Cell and Tissue Research, 2018. Vol. 373. P. 221–232.
66. Gargaglioni L.H., Hartzler L.K., Putnam R.W. The Locus Coeruleus and Central Chemosensitivity // Respir. Physiol. Neurobiol. 2010. Vol. 173, № 3. P. 264–273.
67. D’Adamo M.C. et al. Genetic inactivation of Kcnj16 identifies Kir5.1 as an important determinant of neuronal PCO2/pH sensitivity // J. Biol. Chem. 2011. Vol. 286, № 1. P. 192–198.
68. Sirois J.E. et al. The TASK-1 Two-Pore Domain K+ Channel Is a Molecular Substrate for Neuronal Effects of Inhalation Anesthetics // J. Neurosci. 2000. Vol. 20, № 17. P. 6347–6354.
69. Talley E.M. et al. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons // Neuron. 2000. Vol. 25, № 2. P. 399–410.
70. Nakamura N. et al. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+ ,K+-ATPase // Biochem. J. 1999. Vol. 342, № 2. P. 329–336.
71. Hasselblatt M. et al. Identification of novel diagnostic markers for choroid plexus tumors: A microarray-based approach // Am. J. Surg. Pathol. 2006. Vol. 30, № 1. P. 66–74.
72. Shimura M. et al. Expression and permeation properties of the K+ channel Kir7.1 in the retinal pigment epithelium // J. Physiol. 2001. Vol. 531, № 2. P. 329–346.
73. Kusaka S. et al. Functional Kir7.1 channels localized at the root of apical processes in rat retinal pigment epithelium // J. Physiol. 2001. Vol. 531, № 1. P. 27–36.
74. Cornejo I. et al. Tissue distribution of Kir7.1 inwardly rectifying K+ channel probed in a knock-in mouse expressing a haemagglutinin-tagged protein // Front. Physiol. 2018. Vol. 9, № APR. P. 1–12.
75. Villanueva S. et al. Cleft palate, moderate lung developmental retardation and early postnatal lethality in mice deficient in the Kir7.1 inwardly rectifying K+ channel // PLoS One. 2015. Vol. 10, № 9. P. 1–15.
76. Sergouniotis P.I. et al. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis // Am. J. Hum. Genet. The American Society of Human Genetics, 2011. Vol. 89, № 1. P. 183–190.
77. Pattnaik B.R. et al. A Novel KCNJ13 Nonsense Mutation and Loss of Kir7.1 Channel Function Causes Leber Congenital Amaurosis (LCA16) // Hum. Mutat. 2015. Vol. 36, № 7. P. 720–727.
78. Offermanns S. et al. The potassium channel KCNJ13 is essential for smooth muscle cytoskeletal organization during mouse tracheal tubulogenesis // Nat. Commun. Springer US, 2018. Vol. 9, № 1. P. 1–13.
79. Zhong H. et al. CRISPR-engineered mosaicism rapidly reveals that loss of Kcnj13 function in mice mimics human disease phenotypes // Sci. Rep. 2015. Vol. 5. P. 1–9.
80. Roman D. et al. Conditional loss of Kcnj13 in the retinal pigment epithelium causes photoreceptor degeneration // Exp. Eye Res. Elsevier, 2018. Vol. 176, № June. P. 219–226.
81. Kumar M., Pattnaik B.R. Focus on Kir7.1: Physiology and channelopathy // Channels. 2015. Vol. 8, № 6. P. 488–495.
82. Damkier H.H., Brown P.D., Praetorius J. Cerebrospinal Fluid Secretion by the Choroid Plexus // Physiol. Rev. 2013. Vol. 93, № 4. P. 1847–1892.
83. Iwashita M. et al. Pigment Pattern in jaguar/obelix Zebrafish Is Caused by a Kir7.1 Mutation: Implications for the Regulation of Melanosome Movement // PLoS Genet. 2006. Vol. 2, № 11. P. e197.
84. McCloskey C. et al. The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy // EMBO Mol. Med. 2014. Vol. 6, № 9. P. 1161–1174.
85. Asteria C. Molecular basis of Bartter’s syndrome: New insights into the correlation between genotype and phenotype // Eur. J. Endocrinol. 1997. Vol. 137, № 6. P. 613–615.
86. Peters M. et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies // Am. J. Med. 2002. Vol. 112, № 3. P. 183–190.
87. Feldmann D., Alessandri J.-L., Deschênes G. Large Antenatal Deletion Bartter of the 5 End of the ROMK 1 Gene Causes Syndrome of Antenatal Bartter Syndrome // J. Am. Soc. Nephrol. 1998. Vol. 9, № 12. P. 2357–2359.
88. Simon D. et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. // Nat. Genet. 1996. Vol. 14, № 2. P. 152–156.
89. Cho J.T., Guay-Woodford L.M. Heterozygous Mutations of The Gene for Kir 1.1 (ROMK) in Antenatal Bartter Syndrome Presenting with Transient Hyperkalemia, Evolving to a Benign Course // J. Korean Med. Sci. 2003. Vol. 18, № 1. P. 65–68.
90. Finer G. et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome // J. Pediatr. 2003. Vol. 142, № 3. P. 318–323.
91. Scholl U.I. et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10 // Proc. Natl. Acad. Sci. 2009. Vol. 106, № 14. P. 5842–5847.
92. Bockenhauer D. et al. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations // N. Engl. J. Med. 2009. Vol. 360, № 19. P. 1960–1970.
93. Buono R.J. et al. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility // Epilepsy Res. 2004. Vol. 58, № 2–3. P. 175–183.
94. Baukrowitz T. et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function // Proc. Natl. Acad. Sci. 2010. Vol. 107, № 32. P. 14490–14495.
95. Sala-Rabanal M. et al. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10) // J. Biol. Chem. 2010. Vol. 285, № 46. P. 36040–36048.
96. Yang T. et al. Mutations of KCNJ10 Together with Mutations of SLC26A4 Cause Digenic Nonsyndromic Hearing Loss Associated with Enlarged Vestibular Aqueduct Syndrome // Am. J. Hum. Genet. The American Society of Human Genetics, 2009. Vol. 84, № 5. P. 651–657.
97. Hasan S.M. et al. Lethal digenic mutations in the K+ channels Kir4.1 (KCNJ10) and SLACK (KCNT1) associated with severe-disabling seizures and neurodevelopmental delay // J. Neurophysiol. 2017. Vol. 118. P. 2402–2411.
98. Hejtmancik J.F. et al. Mutations in KCNJ13 Cause Autosomal-Dominant Snowflake Vitreoretinal Degeneration // Am. J. Hum. Genet. 2008. Vol. 82, № 1. P. 174–180.
99. Pattnaik B.R. et al. Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. // PLoS One. 2013. Vol. 8, № 8. P. 1–12.
100. Khan A.O. et al. A distinct vitreo-retinal dystrophy with early-onset cataract from recessive KCNJ13 mutations // Ophthalmic Genet. 2015. Vol. 36, № 1. P. 79–84.
