
Во власти опухоли: паранеопластические поражения ЦНС

Без антирецепторных энцефалитов. Они уже были описаны на Медаче: 1 часть, 2 часть.
▶ Еще по теме: Двигательные нарушения (PDF)
Паранеопластические энцефалиты
Паранеопластические энцефалиты — пока еще плохо изученная тема как в рамках клинического изучения этих синдромов (паранеопластических в целом), так и в фундаментальных аспектах.
Заранее стоит оговориться: одни и те же антитела могут встречаться как в рамках паранеопластического синдрома, так и аутоиммунного. Иногда, в случае, если злокачественное новообразование не дает о себе знать, оно плавно перетекает в рамки аутоиммунного заболевания. Однако стоит понимать, что период от манифестации паранеопластического процесса до выявления самой опухоли может превышать 800 дней. По крайней мере, при отсутствии первичной визуализации опухолевого заболевания, рекомендуется повторять попытки каждые 6 мес в течение 4 лет [1]. Тем не менее, терапия этих синдромов носит один характер — негативная регуляция иммунного ответа.
Структура повествования будет складываться из описания антител, а затем и посиндромальных поражений ЦНС, к которым они могут привести. Для более полного понимания, следует также обращаться к предыдущему обзору паранеопластических и аутоиммунных процессов в рамках двигательных расстройств.
Поражение центральной (в т. ч. и периферической) НС в рамках онкологического заболевания может быть обусловлено тремя более-менее изученными процессами [2]:
- воздействием самой опухоли (как местно-деструктивный рост, так и инфильтрация опухолевых клеток в случае, например, лимфомы);
- идиосинкратическими и изоиммунными явлениями (в рамках амилоидоза, моноклональной гаммапатии неясного генеза, криоглобулинемического васкулита, секреции цитокинов при POEMS-синдроме);
- непосредственно аутоиммунным процессом (далее он будет называться паранеопластическим) с образованием антител к экспрессирующимся опухолью антигенам, подобным белкам собственного организма, среди которых выделяют:
а) мембранные антигены — те, что находятся на поверхности мембраны (в основном, рецепторы, каналы);
б) внутриклеточные — думаю, догадываетесь, где они находятся.
Патогенез
Описание индуцированного паранеопластического процесса проще раскрыть в рамках неврологических иммунно-индуцированных побочных эффектов, в случае использования чекпоинт-ингибиторов — препаратов, усиливающих противоопухолевую иммунную активность (рис.1). Побочные эффекты раскрывают вероятные механизмы развития как аутоиммунной реакции, так и индукцию либо усиление паранеопластического процесса [3].
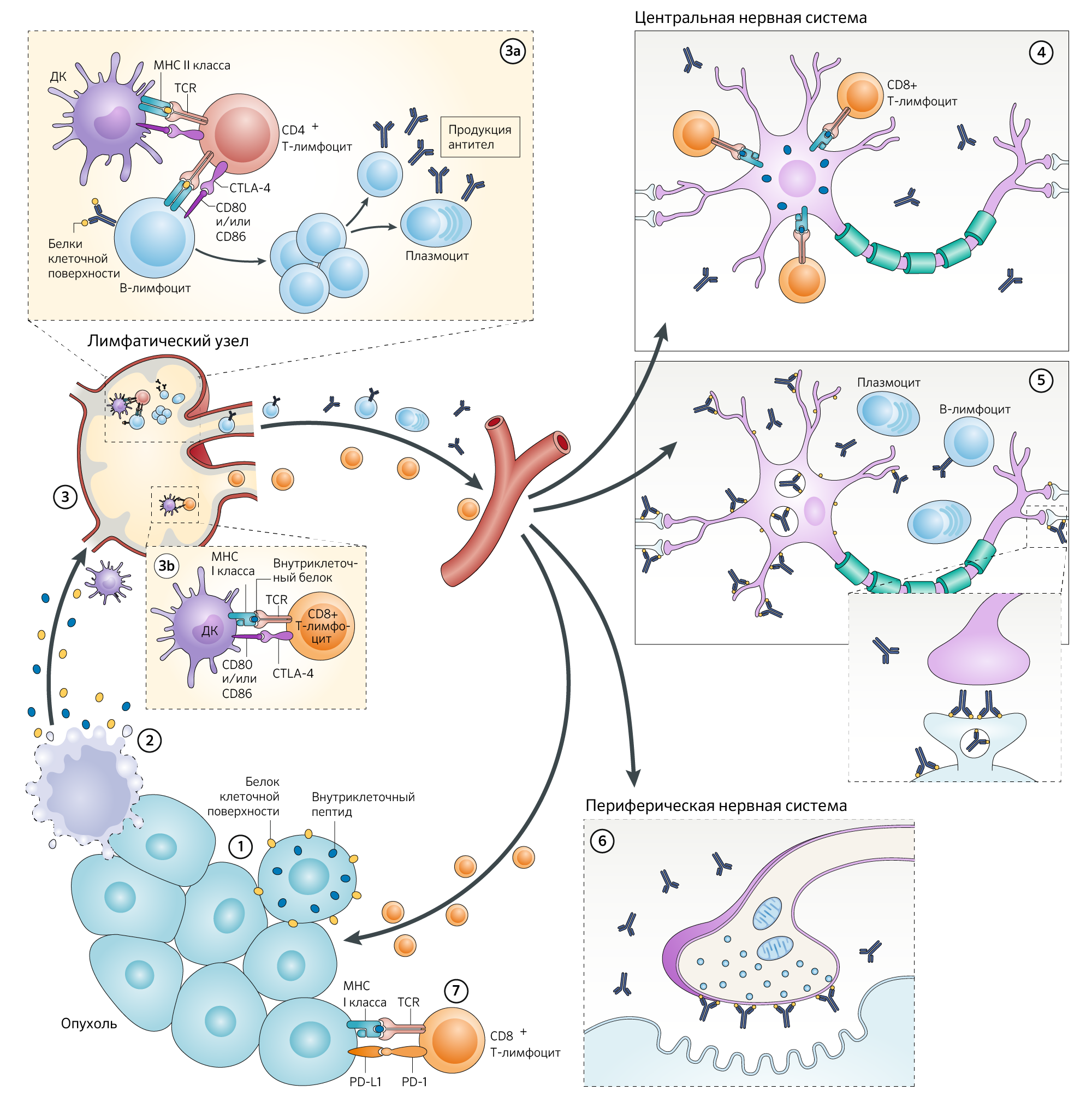
Рисунок 1. Предполагаемые механизмы развития ПНС в рамках формирования неврологических иммунно-индуцированных побочных эффектов в ответ на чекпоинт-ингибиторы.
Статьи, раскрывающие механизмы работы чекпоинт-ингибиторов, мириады взаимодействий опухоли и ее окружения с иммунной системой, вы можете прочесть на нашем портале по ссылкам pH-чувствительные антитела к CTLA-4, Ингибирование Chk1, Новые рычаги опухолевого сопротивления, Клеточная система таргетной доставки анти-PD-1 антител, Ингибиторы контрольных точек для лечения рака головы и шеи, Контрольные точки и их ингибиторы.
На рисунке показан многоступенчатый процесс, в результате которого формируются иммуноопосредованные паранеопластические заболевания, поражающие центральную и периферическую нервные системы.
Внутриклеточные или мембранные нейронные белки, экспрессируемые в опухоли (1), высвобождаются при гибели опухолевой клетки (2).
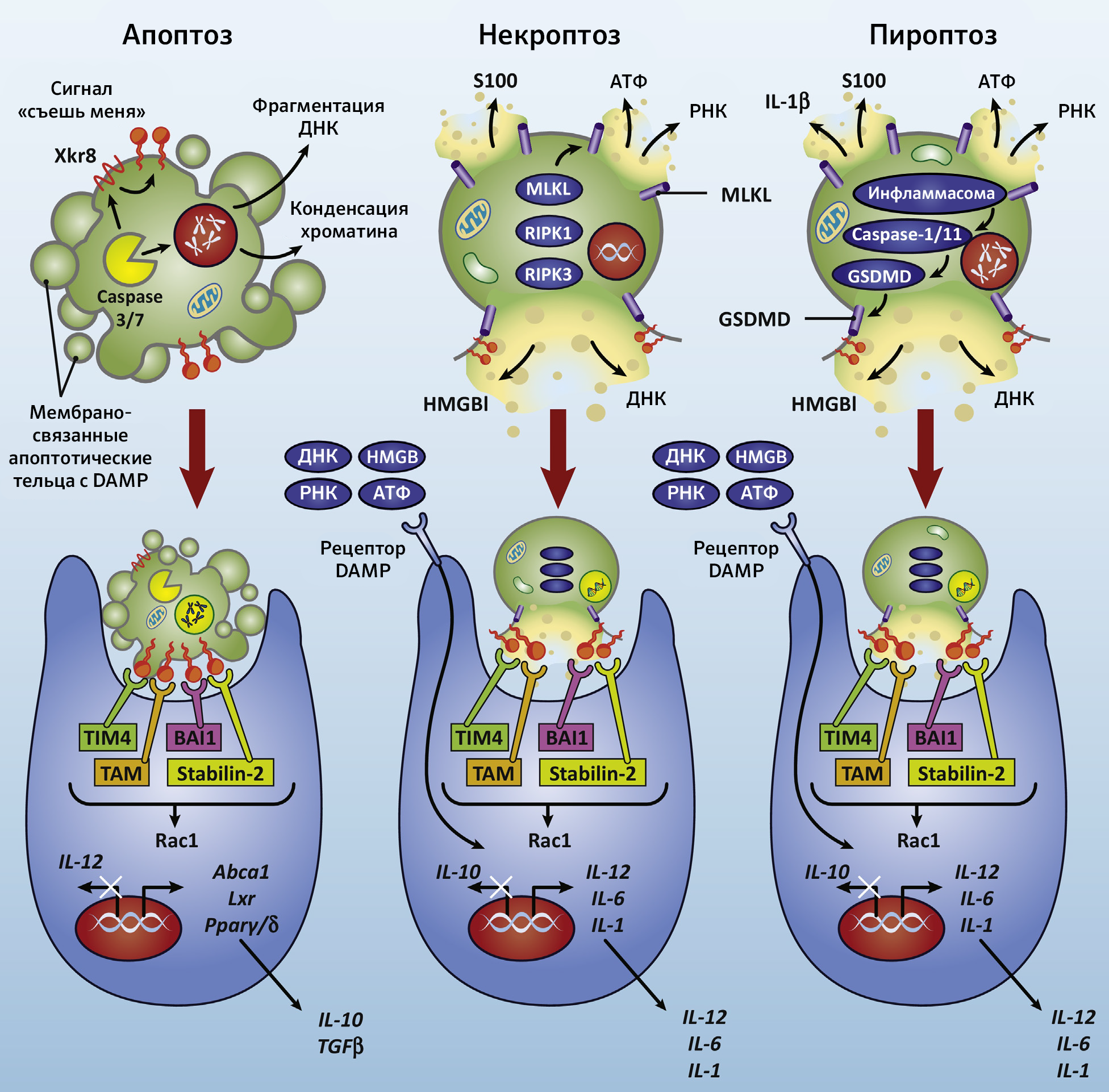
Вряд ли клеточная гибель связана с апоптозом, ибо в этом случае конечными медиаторами являются IL-10 и TGFb, которые, по сути, противовоспалительные. Предполагается, что фагоцитоз апоптотических телец обуславливает иммуносупрессию и развитие опухолевого окружения. В то же время, фагоцитоз клеточных элементов, погибших при несколько иных обстоятельствах, способствует секреции макрофагами провоспалительных цитокинов: IL-12, IL-6, IL-1 (рис. 2). Выраженный провоспалительный эффект может оказаться тем необходимым шагом на пути к аутоиммунному процессу [4].
Для простоты все внутриклеточные онконейрональные белки или пептиды представлены в виде синих округлых форм, а все поверхностные нейрональные белки и рецепторы показаны желтыми округлыми формами. Эти белки захватываются и обрабатываются дендритными клетками (ДК) и презентируются в виде антигенов в регионарных лимфатических узлах представителям адаптивной иммунной системы (3). Механизмы развития паранеопластического синдрома (ПНС), обусловленные мембранными антигенами, преимущественно опосредуются В-клетками и аутоантителами. В лимфатическом узле возникают сложные взаимодействия между В-клетками и CD4+ Т-хелперами, которые были активированы путем связывания нейронального антигена непосредственно с родственным рецептором В-клеток и косвенно с рецептором Т-клеток (TCR) в контексте молекул MHC класса II, экспрессируемых на антигенпрезентирующих клетках (DC), что, соответственно, приводит к пролиферации В-клеток и выработке плазматических клеток и B-клеток памяти (3а).
Факты указывают на то, что В-клетки, секретирующие антитела, достигают ЦНС, где они подвергаются повторной стимуляции, антиген-зависимому созреванию аффинности, клональной экспансии и дифференцировке в плазматические клетки с дальнейшим продуцированием аутоантител. В ЦНС эти аутоантитела связываются со своими мишенями на клеточной поверхности, таким образом, вызывая структурные и функциональные изменения, которые, в зависимости от цели, приводят к различным формам паранеопластического энцефалита (например, лимбический энцефалит, связанный с аутоантителами, нацеленными на рецепторы α-амино-3-гидрокси-5-метил-4-изоксазолепропионовой кислоты [AMPAR]) (5). Если антиген клеточной поверхности экспрессируется в периферической нервной системе, где отсутствует гематоэнцефалический барьер, аутоантитела достигают цели напрямую, что приводит к неврологическому расстройству (например, синдром Ламберта-Итона, связанный с аутоантителами к потенциал-зависимым кальциевым каналам, экспрессирующимся на пресинаптической мембране в нервно-мышечных синапсах) (6). Напротив, ПНС, обусловленные внутриклеточными нейрональными антигенами (также называемыми онконейрональными антигенами) формируются в результате агрессии CD8+ цитотоксических Т-лимфоцитов (CTL), которые активируются антигенами, распознаваемыми в контексте молекул MHC класса I, представленных на DC (3b).
Проникновение CTL в ЦНС приводит к гибели нейронов (4); данное явление часто сопровождается обнаружением аутоантител к мишеням-антигенам CTL. Однако эти аутоантитела, по-видимому, не играют главной роли в патогенезе. Та же цитотоксическая активность CD8+ лимфоцитов обладает противоопухолевым эффектом (7), что, вероятно, объясняет наблюдение о том, что опухолевые заболевания у пациентов с ПНС часто имеют скорее вялотекущее течение, по сравнению с пациентами без ПНС. Пока неизвестно, имеют ли аутоантитела, нацеленные на антигены клеточной поверхности, клинически соответствующие противоопухолевые эффекты.
Чекпоинт ингибиторы (ICI), блокирующие CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) (3a и 3b) или взаимодействие PD-1 (programmed death cell-1) с PD-L1 (programmed death cell ligand -1) (7), усиливают противоопухолевые иммунные ответы, которые, при их направленности против антигенов, экспрессирующихся в ЦНС или периферической нервной системе, могут приводить к проявлению ПНС в виде неврологических иммунных побочных эффектов.
Говорить мы будем о поражениях ЦНС, обусловленных онконейрональными антителами, то бишь, антителами к внутриклеточным антигенам. Как уже было описано выше, за развитие паранеопластического процесса в равной степени играют как гуморальный, так и клеточный иммунные ответы. Представление онконейронального антигена может происходить двумя путями:
- В результате мутагенного процесса в самой опухоли с последующим высвобождением этого антигена после ее гибели.
- По причине нарушения ГЭБ, местно-деструктивного эффекта опухоли, поражения нейронов, их гибели и опять же высвобождения антигенов.
Итак, начнем.
Анти-Hu антигены
Так называемые анти-Hu антитела образуются в ответ на три белка, экспрессирующихся в ЦНС: HuD, HuC, HuB [5].
Hu — семейство внутриклеточных белков, в которое кроме описанных трех белков входит и 4-й — HuA, но из-за практически повсеместной экспрессии он не рассматривается как мишень в рамках развития паранеопластического синдрома. Это семейство белков играет ключевую роль в нейрональном развитии, выступая в качестве нейрон-специфических РНК-связывающих белков. Также было продемонстрировано, что HuD способствует стабилизации мРНК, кодирующей BDNF (brain-derived neurotrophic factor, нейротропный фактор роста). HuD в основном экспрессируется в ядре, но может мигрировать в цитоплазму, где располагается в небольших гранулах. Цитоплазматическая локализация HuD, по-видимому, необходима для дифференцировки нейронов. Кроме того, было показано, что HuD взаимодействует с протеинкиназой B-альфа (AKT1), активируя рост дендритов или нейритов. Таким образом, белки Hu, в частности HuD, оказывают большое влияние на множество ключевых нейронных процессов.
Прежде чем перейти к клинико-диагностическим аспектам, следует упомянуть, что иногда в литературе упоминается другое название анти-Hu антител — ANNA-1 (антинейрональные ядерные антитела первого типа).
Чаще всего анти-Hu антитела при паранеопластическом синдроме определяются среди лиц, больных мелкоклеточным раком легких (МКРЛ) (около 85 % случаев), реже при злокачественных новообразованиях кишечника, простаты, молочной железы, мочевого пузыря, яичников, семиноме, тимоме, лимфоме и т. д.
В исследовании 200 анти-Hu-положительных случаев опухоль не была обнаружена у 33 пациентов [6].
Клиника
Клинические проявления заболевания проще всего раскрыть на примере клинических случаев:
Случай первый
55-летняя женщина, поступила с жалобами на быстро прогрессирующую потерю памяти [7]. Она не могла вспомнить, что происходило в течение прошлых пяти дней, а также могла начать говорить вне контекста. Также пациентка страдала тяжелой депрессией. В неврологическом статусе, кроме нарушения памяти, патологии выявлено не было. Данные опросников: Краткая шкала оценки психического статуса (MMSE) — 18/30, Шкала оценки инструментальной деятельности в повседневной жизни (IADL) — 1,7.
Результаты МРТ головного мозга с контрастированием не выявили патологии. ЭЭГ показало транзиторное двустороннее замедление волн в лобно-височных областях. Клинический анализ крови, мочи, биохимия крови, исследование функции щитовидной железы, антитела к ВИЧ, экспресс-тест на реагин плазмы, тесты на свертываемость, уровни витамина B12 и фолиевой кислоты в сыворотке были в пределах нормы. Исследования цереброспинальной жидкости (ЦСЖ) выявили умеренный плеоцитоз с преобладанием лимфоцитов и умеренное повышение уровня белка до 45,6 мг/дл.
Результаты исследования ликвора и сыворотки крови на инфекции были отрицательны. При поиске антител на предмет выявления системных, аутоиммунных и паранеопластических заболеваний в сыворотке крови были обнаружены анти-Hu антитела.
Дальнейшая диагностика позволила установить причину развития паранеопластического синдрома — плоскоклеточная опухоль шейки матки (рис. 3).
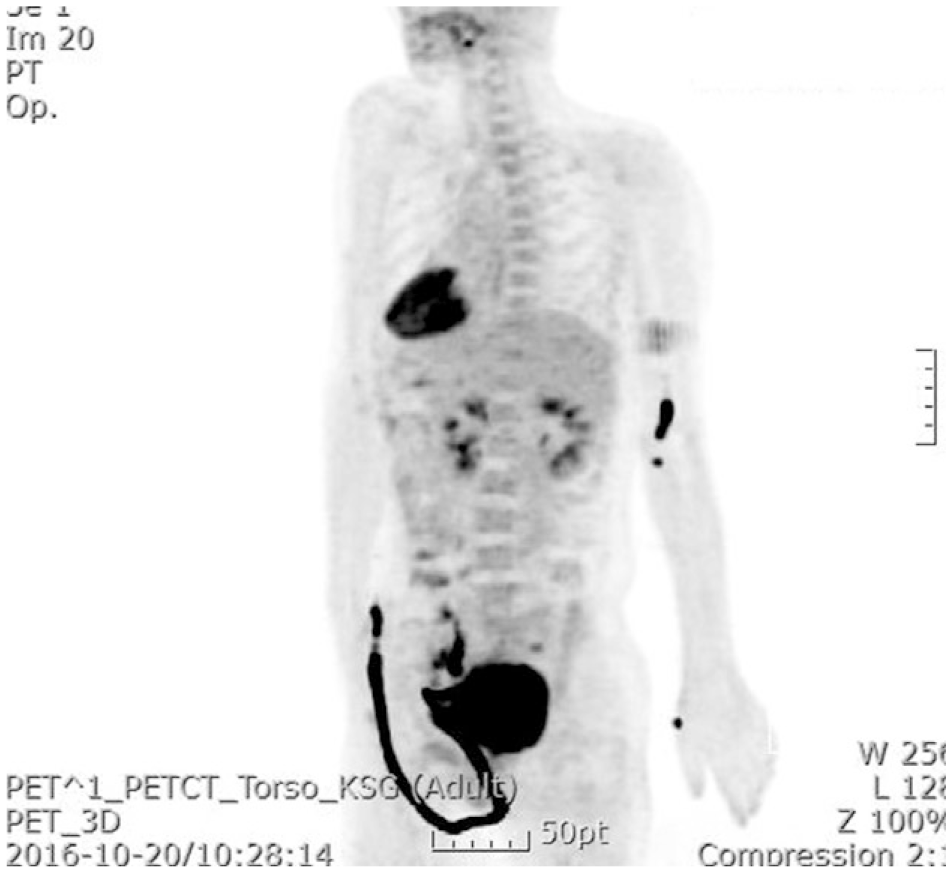
Рисунок 3. Исследование ПЭТ-КТ выявило интенсивное поглощение 18F-фтордезоксиглюкозы в шейке матки, а также увеличенные лимфатические узлы с поглощением ФДГ в боковых стенках таза.
Учитывая наличие паранеопластического лимбического энцефалита:
- клиника острого нарушения памяти и психического состояния в виде тяжелой депрессии;
- замедление волн во фронтотемпоральных областях на ЭЭГ;
- небольшой плеоцитоз, альбумино-клеточная диссоциация;
- выявление анти-Hu антител в сыворотке крови;
совместно с химиолучевой терапией, проводилось лечение высокими дозами стероидов с постепенным их снижением в течение 8 недель. Пациентка также лечилась антидепрессантами, антипсихотиками и ингибиторами ацетилхолинэстеразы. Тем не менее, эффект оставлял желать лучшего. После 1 года терапии выраженность депрессии снизилась, и показатель MMSE улучшился с 18/30 до 22/30.
Случай второй
Редчайший случай мимикрии под болезнь моторных нейронов [8]. 64-летняя женщина с сахарным диабетом 2 типа (HbA1C: 6,6 %) страдала от поясничных болей с иррадиацией вниз по правой ноге с периодическим ощущением покалывания и онемения, сопровождающихся слабостью в конечности и падениями. Через два месяца от манифестации заболевания ей требовались ходунки для преодоления больших дистанций. В течение последующих 5 месяцев она испытывала медленно прогрессирующую слабость в правой ноге. Неврологическое обследование выявило вялый парез в проекции L3-S1 справа. Рефлексы оживлены (3+), за исключением отсутствующих правого подколенного и ахиллова с двух сторон. Подошвенные рефлексы — сгибательные. Также выявлялось нарушение чувствительности в дистальных отделах нижних конечностей.
ЭНМГ показало сниженный моторный ответ со всех волокон правой ноги без воздействия на проводимость сигнала, отсутствие блоков проводимости или признаков демиелинизации. Сенсорный ответ с обеих ног отсутствовал. Игольчатая ЭНМГ выявила начальный этап денервации в проекции люмбосакральных и грудных миотомов справа.
Учитывая асимметричный периферический парез, фасцикуляции и потерю чувствительности, формируется представление о поражении мотонейронов люмбосакрального отдела и периферических нервов. В первую очередь, была рассмотрена радикулоплексопатия справа (диабетическая амиотрофия) с диабетической полинейропатией. Однако денервация волокон, распространяющаяся на грудные миотомы и клинически здоровую ногу, опровергали диагноз унилатеральной радикулоплексопатии. Более того, хотя манифестация заболевания и напоминает в большей степени клинику поражения именно нижнего мотонейрона (НМН), патологически высокие рефлексы предполагают раннее вовлечение в заболевание верхнего мотонейрона (ВМН).
Несмотря на то, что сенсорный дефицит и отсутствие парезов на других конечностях являются атипичными, все же присутствие вовлечения в патогенез как НМН, так и ВМН наталкивает на диагностирование бокового амиотрофического склероза (БАС), а именно его подтипа с доминирующим вовлечением НМН + диабетической полинейропатии, которая объясняет сенсорный дефицит. Дифференциальная диагностика также включает моторною нейронопатию и моторную нейропатию, среди которых встречаются мультифокальная моторная нейропатия и мультифокальная приобретенная моторная аксонопатия (+ диабетическая полинейропатия). Лабораторные исследования продемонстрировали нормальные уровни B12, церулоплазмина, ТТГ, отсутствие повышения провоспалительных маркеров и т. д. КФК была незначительно повышена, что может наблюдаться при болезни моторного нейрона. МРТ без контраста выявило спондилолистез L5–S1.
В течение последующих 4 месяцев состояние пациентки оставалось стабильным. Однако 11 месяцев спустя манифестации заболевания проявились слабость верхних конечностей, диплопия, одышка и дисфагия с потерей 25 фунтов веса в течение пяти недель. Отмечалась бинокулярная горизонтальная диплопия с прогрессией до полного пареза горизонтального взора. Также отмечались онемение и покалывание левой ноги и рук. Никаких когнитивных нарушений, судорожных припадков или лихорадки не было.
У пациентки острота зрения была сохранена, патологии со стороны полей зрения выявлено не было. Конвергенция и вертикальный взор интактны, однако горизонтальный взор отсутствовал с двух сторон, что не было обусловлено подавлением вестибуло-окулярного рефлекса. Присутствовал периферический парез лицевого нерва. Небо симметрично, занавеска не провисала, язык двигался нормально, без фасцикуляций. Дизартрия. Нижнечелюстные рефлексы оживлены. Объем квадрицепса справа уменьшился, появились фасцикуляции. Легкий вялый парез отмечен в сгибателях шеи, дельтовидных мышцах и двуглавой мышце с обеих сторон.
Вялый парез правой нижней конечности. Рефлексы в верхних конечностях оставались патологически высокими, в нижних — отсутствовали. Левый подошвенный рефлекс — сгибательный, справа — отсутствует. Снизилась вибрационная чувствительность в дистальных отделах нижних конечностей.
Перейдем к нейровизуализации. T2-FLAIR демонстрирует гиперинтенсивный сигнал с дорсальной части моста, вовлекая область лицевых холмиков и ядра VI ЧН (рис. 4, A и B). Гиперинтенсивный сигнал проходит на всем протяжении вплоть до ростральной части моста, что указано стрелочками на изображениях C и D (рис. 4).
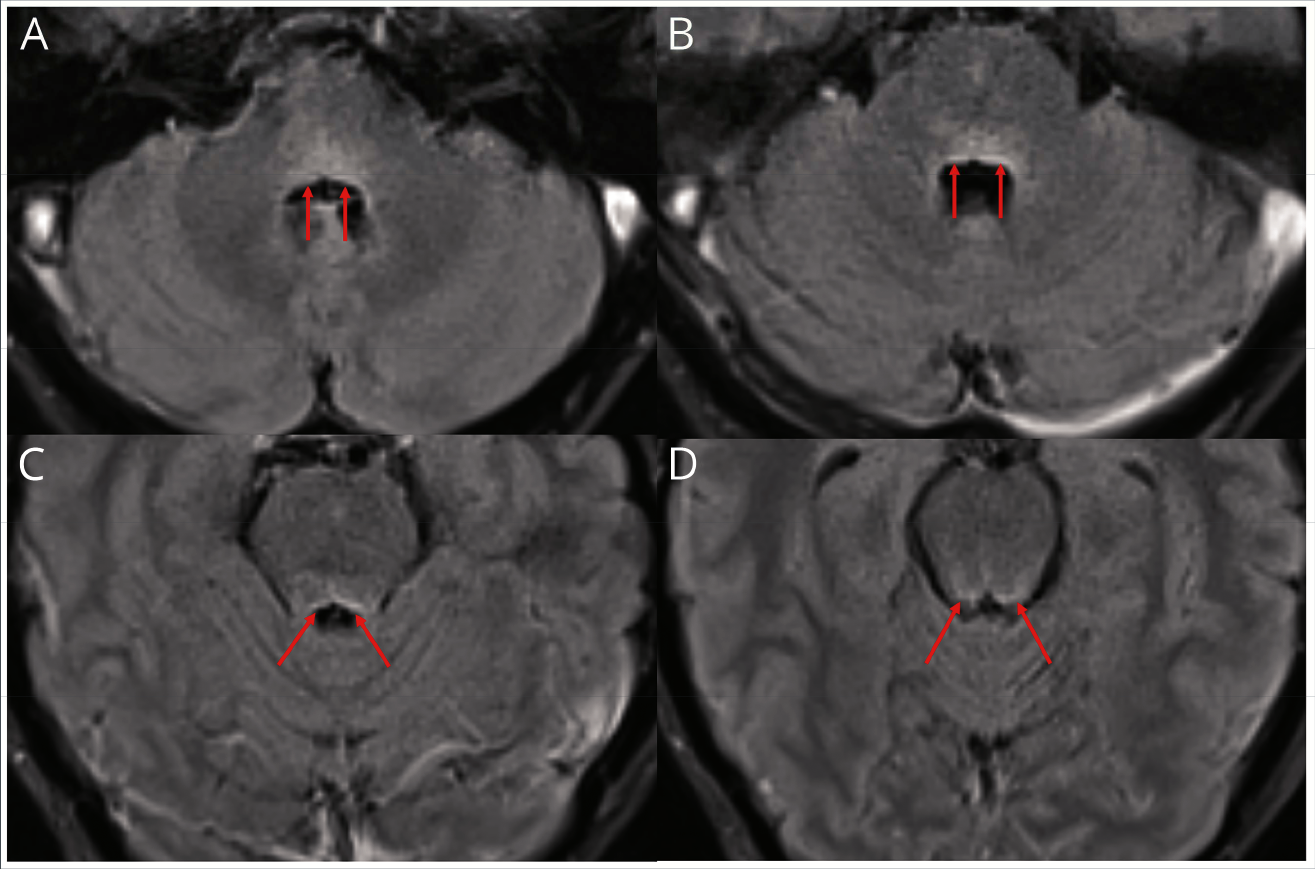
Рисунок 4. МРТ Т2 FLAIR в аксиальной проекции (описание в тексте).
Хотя офтальмоплегия и может сопровождать бульбарную форму БАС, а глазодвигательные расстройства наблюдаются среди пациентов, находящихся на ИВЛ, тем не менее, офтальмоплегия при БАС скорее уж казуистика. Однако наличие дисфагии и потери веса может наталкивать на мысль о присоединении энцефалопатии Вернике, объясняющей офтальмоплегию, что тут же опровергается прогрессированием заболевания, несмотря на большие дозы тиамина.
Прогрессирующий супрануклеарный парез и мультисистемная атрофия могут редко идти рука об руку с заболеваниями мотонейрона, но отсутствие нарушения вертикального взора, паркинсонизма, автономной дисфункции и атаксии не согласуется с ПСП и МСА.
Хотя ЭНМГ и показало вовлечение в основном НМН, скорость прогрессирования, в т. ч. и сенсорной симптоматики, наличие офтальмоплегии и гиперинтенсивного сигнала с моста вынуждают отречься от диагноза БАС в пользу заболевания другой этиологии — воспалительной, паранеопластической или аутоиммунной — вовлекающего мотонейроны и ядра черепных нервов.
КТ грудной клетки, брюшной полости и таза, трансвагинальное УЗИ не выявили признаков злокачественного новообразования или лимфаденопатии. Анализ ЦСЖ выявил уровень белка 503 мг/л, но в остальном цитология, цитометрия и серологические тесты на анти-GQ1b, анти-GM1, болезнь Лайма и антитела к другим вирусным возбудителям, а также бактериологическое исследование ликвора были отрицательными. Повторная ЭНМГ выявила вновь возникшую неоднородную, не зависящую от длины волокон потерю сенсорных ответов в левой верхней конечности. Высокие дозы метилпреднизолона и иммуноглобулинов в/в оказались неэффективны. В дальнейшем прогрессировали дыхательные расстройства, что привело к дезориентации и летаргии. ЭЭГ не выявила судорог, повторная МРТ головного мозга была без изменений. Пациент продолжал ухудшаться и в конце концов умер от дыхательной недостаточности. Позже результаты ликвора и крови на антитела продемонстрировали анти-Hu-антитела.
Обсуждение
Диагностика атипичных форм моторных нейропатий и НМН-доминантных БАС является настоящим испытанием для неврологов. Отсутствие качественных маркеров заболевания приводит к выставлению неверного диагноза БАС примерно в 10 % случаев. Шейная спондилогенная миелорадикулопатия, полирадикулопатия, МСА и мультифокальная моторная нейропатия иногда ошибочно трактуются как БАС. Особую сложность представляет ранняя диагностика заболевания, когда проявления поражений НМН и ВМН еще не распространены по всему телу.
Изначально клинические данные вовлечения НМН и, в меньшей степени, ВМН на ранних этапах течения болезни нашего пациента, а также прогрессирующая денервации в множестве грудных и пояснично-крестцовых миотомах удовлетворяли пересмотренным критериям Эль-Эскориал для диагностики вероятного БАС.
В редких случаях паранеопластические расстройства могут вызывать болезнь моторных нейронов и маскироваться под БАС, чаще всего ассоциированной с Hu2-антителами. Паранеопластический синдром БМН может проявляться либо только синдромом НМН в 63 % случаев, либо поражением и НМН, и ВМН в 37 % случаев. Паранеопластический синдром БМН следует подозревать у лиц моложе 40 лет с известным злокачественным новообразованием, быстрым прогрессированием, подострым началом, вовлечением экстрамоторных нейронов, моноклональной гаммапатией, лимфаденопатией или повышенными маркерами воспаления.
Быстро прогрессирующий выраженный парез в правой ноге без клинического поражения левой ноги нетипичен для БАС. Диабет может объяснить начальные дистальные сенсорные симптомы, но манифестация частичного, ассиметричного сенсорного дефицита демонстрирует еще один красный флаг вовлечения экстрамоторных нейронов, вовлекающих ганглии задних корешков, вероятно, вторично по отношению к анти-Hu. Когда состояние пациентки с развитием офтальмоплегии быстро ухудшилось, стало ясно, что у нее нет БАС.
Это первый зарегистрированный случай офтальмоплегии, связанной с анти-Hu-антителами, и БМН. Анти-Hu-ассоциированная БМН впервые была описана в 1995 году в контексте МКРЛ с поражением нейронов передних рогов, клеток Пуркинье и нейрогенной денервацией. Клинический спектр паранеопластических расстройств, связанных с анти-Hu, с тех пор расширился, включив лимбический и стволовой энцефалиты, энцефаломиелит, сенсорную нейронопатию, синдром Ламберта — Итона и опсоклонус-миоклонус синдром. Недавно сообщалось об офтальмоплегии у пациента с липосаркомой и антителами против Hu, но без проявления БМН.
В случае развития атипичных форм БМН следует проводить качественный скрининг на наличие паранеопластического синдрома, особенно, учитывая возможность его регресса на фоне терапии злокачественного новообразования либо стабилизации при использовании иммунотерапии.
Анти-Ма1/Ма2 паранеопластические энцефалиты
В исследовании P. Schwenkenbecher, et al 18-ти пациентов с паранеопластическим синдромом, обусловленным анти-Hu антителами, выявили следующие клинические паттерны [9].
- Периферическая нейропатия у восьми пациентов.
- Ромбэнцефалит с мозжечковой и/или стволовой симптоматикой у семи пациентов:
a) у четырех пациентов — мозжечковая атаксия, дизартрия, дисфагия, гемипарез, глазодвигательные расстройства (парез отводящего нерва, межъядерная офтальмоплегия), нарушение саккад и/или нистагм;
b) у трех пациентов — только стволовая симптоматика: глазодвигательные расстройства (парез отводящего нерва), гемипарез и гемигипестезия;
c) у одного пациента в дополнение к стволовому энцефалиту на МРТ был обнаружен участок поражения в шейном отделе. Симптоматика проявлялась только гемигипестезией лица и тела;
d) у остальных шести пациентов симптоматика складывалась из центрального и периферического компонентов поражения, проявляясь как сенсорная с прогрессированием до сенсомоторной полинейропатии дистальных отделов рук и ног. Так что не удивляйтесь, если пациент поступит изначально с синдромом поражения ЦНС, а затем постепенно (или же практически за день) будет прогрессировать до смешанной патологии ЦНС + ПНС.
Кроме того, у пациентов с ромбэнцефалитом может наблюдаться вертиго. Редким проявлением считается экстрапирамидная симптоматика в виде тремора [10]. - Лимбический энцефалит — у трех пациентов, клинически демонстрировавших поведенческие нарушения (агрессию, тревожность), дезориентацию, мнестический синдром. Двое пациентов страдали от фокальных эпилептических припадков. У одного из них развились генерализованные припадки с переходом в эпистатус. Кроме того, лимбический энцефалит может проявиться спутанностью сознания с одновременным развитием параноидного бреда [11].
- Миелит — как в рамках энцефаломиелита, так и поперечного протяженного миелита [12].
Довольно редким проявлением можно считать развитие опсоклонус-миоклонус синдрома (ОМС), который чаще всего ассоциирован с нейробластомой у детей [13] и МКРЛ у взрослых пациентов [14]. Однако стоит ли проводить прямую ассоциацию между анти-Hu и ОМС? По крайней мере, между данными явлениями нет четко установленной причинно-следственной связи, а наличие анти-Hu синдрома скорее характеризует развитие лимбического энцефалита на фоне либо после регресса ОМС. ОМС наблюдается среди пациентов, у которых наличествуют несколько антител (анти-Hu, анти-Ri и т. д.), что не исключает вовлечения антител к Hu в развитие этого синдрома [15].
Впрочем, этого не скажешь об анти-Ma2-энцефалите, где клиника ОМС соответствует выявлению онконевральных антител в сыворотке крови и ликворе.
Поэтому далее поговорим про анти-Ма-антитела.
К онконевральным белкам Ма относятся три белка, среди которых только Ма2 экспрессируется селективно в мозговой ткани; Ма1 экспрессируется еще и в половых клетках, тогда как Ма3 — почти повсеместно, что затрудняет его использование в качестве онконеврального антигена при диагностике заболеваний (а также и то, что антитела к нему экспрессируются совместно с антителами к другим Ма-белкам при паранеопластическом заболевании с одним и тем же эпитопом с Ма1) [16].
Анти-Hu и анти-Ma паранеопластические синдромы проявляются мириадами клинических неврологических симптомов, включающих проявления со стороны как ПНС, так и ЦНС. Однако, если в случае с анти-Hu, онкопоиск в первую очередь должен быть направлен на выявление опухолевых заболеваний легких, для анти-Ма в этом случае обязательно начать с поиска герминогенных опухолей.
Приведем далее перевод двух клинических случаев одного заболевания: мужчины 71 года и девушки 16 лет:
Первый клинический случай [17]
Мужчина, 71 год, обратился в клинику по поводу головокружения, атаксии и вертиго. Из анамнеза известно злоупотребление алкоголем и артериальная гипертензия.
Неврологическое обследование выявило быстрые неритмичные саккадические разнонаправленные движения глаз (опсоклонус, см. Видео); атаксию, дизартрию, акционный тремор и миоклонус проксимальных отделов ног.
Изначальные лабораторные исследования, включая определение уровня тиамина, не выявили аномалий.
Анализ спинномозговой жидкости: цитоз 8 кл/мкл (контрольное значение ≤ 5), белок 88 мг/дцл (контрольное значение ≤ 35) и 5 олигоклональных полос (контрольное значение < 4); посев на инфекцию отрицателен. Исследование крови на антитела продемонстрировало наличие анти-Ma2 IgG.
МРТ головного мозга: без патологии.
ПЭТ с фтордезоксиглюкозой: никаких признаков онкологии не обнаружено.
Исходя из неврологического осмотра и лабораторных данных, был выставлен диагноз «Синдром опсоклонус-миоклонус на фоне анти-Ma2 энцефалита ствола головного мозга».
Синдром опсоклонус-миоклонус состоит из опсоклонуса, миоклонических подергиваний и атаксии и может быть связан с аутоиммунными или паранеопластическими заболеваниями, поражающими ствол мозга у взрослых.
Терапия глюкокортикоидами, ритуксимабом и плазмаферезом привела к уменьшению опсоклонуса и миоклонуса, однако в дальнейшем атаксия прогрессировала, и, несмотря на дополнительную терапию циклофосфамидом, через год пациенту пришлось пользоваться инвалидным креслом.
Второй клинический случай [18]
Девушка, 16 лет, поступила в отделение неотложной помощи с вертиго, беспокоящим ее в течение одной недели. Со слов, головокружение началось внезапно, по типу ощущения вращения в пространстве. Далее присоединились головная боль, тошнота и нарушения походки.
Из анамнеза известно, что на второй день заболевания ее лечащий врач прописал 5-дневный курс приема преднизона и меклизина (антигистаминный препарат) дважды в день. Терапия не привела к улучшению, что потребовало проведения МРТ головного мозга. Никаких патологий, кроме синусита, выявлено не было. Доза меклизина была увеличена, а также добавлен цефдинир (цефалоспорин III поколения) с целью лечения синусита, однако симптомы продолжали ухудшаться. На момент обращения в клинику пациентка не могла стоять из-за головокружения. Она жаловалась на головную боль и тошноту, при осмотре выявлялись нистагм, атаксия и дисметрия. Примерно за год до обращения пациентка испытала один эпизод головной боли, но в остальном была здорова. Ее семейный анамнез включал рассеянный склероз у бабушки и мутацию фактора Лейдена и протромбина у брата.
Дифференциальная диагностика проводилась между инфекционными заболеваниями (в том числе, менингитом), аутоиммунными/паранеопластическими энцефалитами, системными аутоиммунными заболеваниями, токсическими/метаболическими нарушениями (в т. ч. отравлением, дефицитом витаминов B1 и B12) и сосудистой патологией (например, тромбозом венозного синуса).
МР-ангиография и венография головы и шеи не выходили за пределы физиологической нормы, не считая ранее описанного синусита, что исключило вероятность тромбоза и артериальной диссекции.
Анализ ликвора выявил небольшой плеоцитоз с преобладанием лимфоцитов (11 лейкоцитов на мкл, нейтрофилов — 0 %, лимфоцитов — 78 %, моноцитов — 2 %, недифференцированных клеток — 20 %, эритроцитов — 0; белок, глюкоза в норме; исследование на инфекции результатов не дало).
Был приглашен офтальмолог с целью исключения оптического неврита. Оптический неврит не был выявлен, однако аномальные движения глаз, ранее описанные как нистагм, оказались опсоклонусом.
Кроме того, мать пациентки описала недавнее появление подергивания конечностей во время сна, тем самым наведя на предположение о синдроме опсоклонус-миоклонус.
ЭЭГ не показало эпилептиформной активности. С целью обследования на паранеопластическую этиологию был взят анализ мочи на исследование уровней секреции ванилилминдальной и гомованилиновой кислот (для исключения нейробластомы). Хотя результаты оказались неоднозначными, нейробластома в возрастной группе пациентки проявляется очень редко.
Также была проведена МРТ органов грудной клетки, брюшной полости и таза для дальнейшей оценки паранеопластической причины ее симптомов.
МРТ органов малого таза выявила тератому левого яичника диаметром 3,5 см (рис. 5).
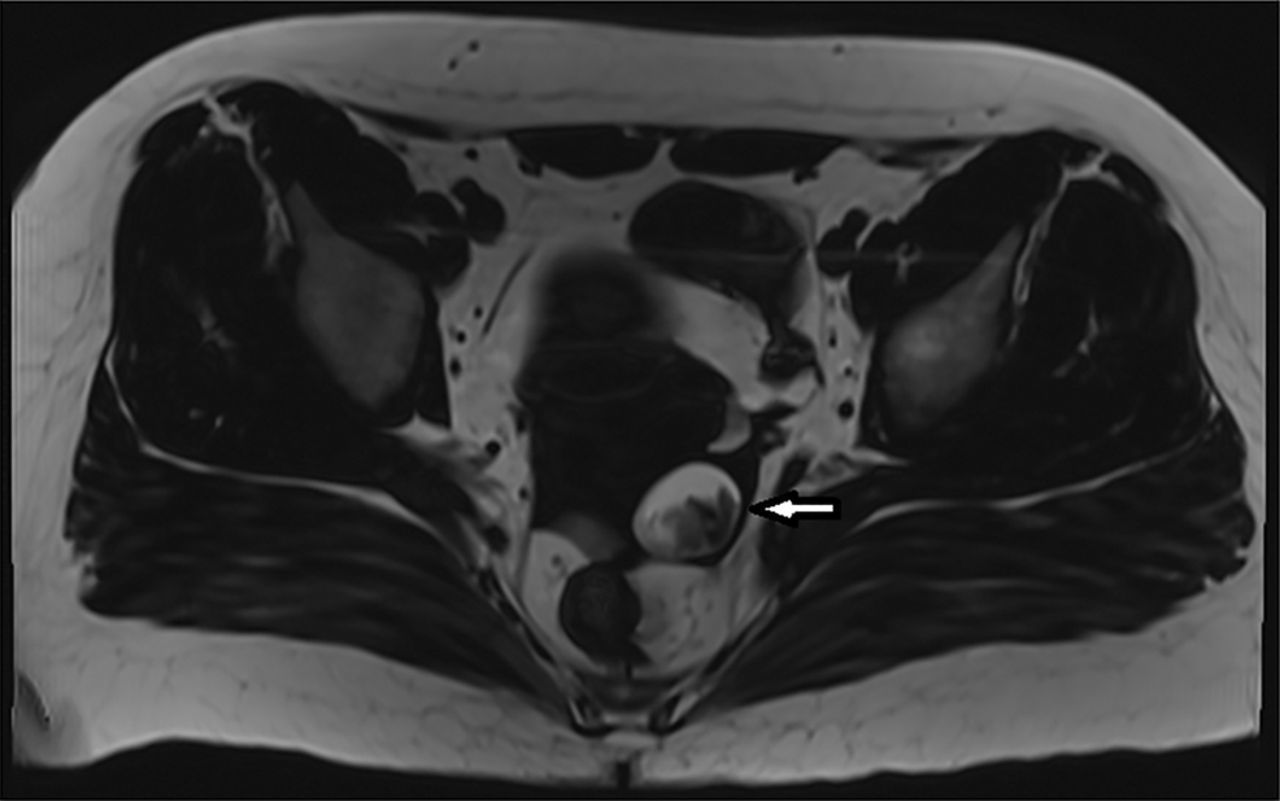
Рисунок 5. МРТ органов малого таза с тератомой левого яичника.
Проведена цистэктомия левого яичника с последующей верификацией образования как зрелой кистозной тератомой яичника.
Из объемного спектра исследуемых антител в ликворе и крови был выявлен повышенный титр антител к GAD65 в сыворотке (0,10 нмоль/л, в норме ≤ 0,02 нмоль/л). Головокружение и миоклонус постепенно регрессировали, пациентка провела две недели в реабилитационном отделении.
Через две недели после выписки у пациентки снова развилось ухудшение опсоклонуса и миоклонуса, а также поведенческие нарушения. Повторная госпитализация и визуализация брюшной полости и таза выявила рецидив тератомы яичника (рис. 6).
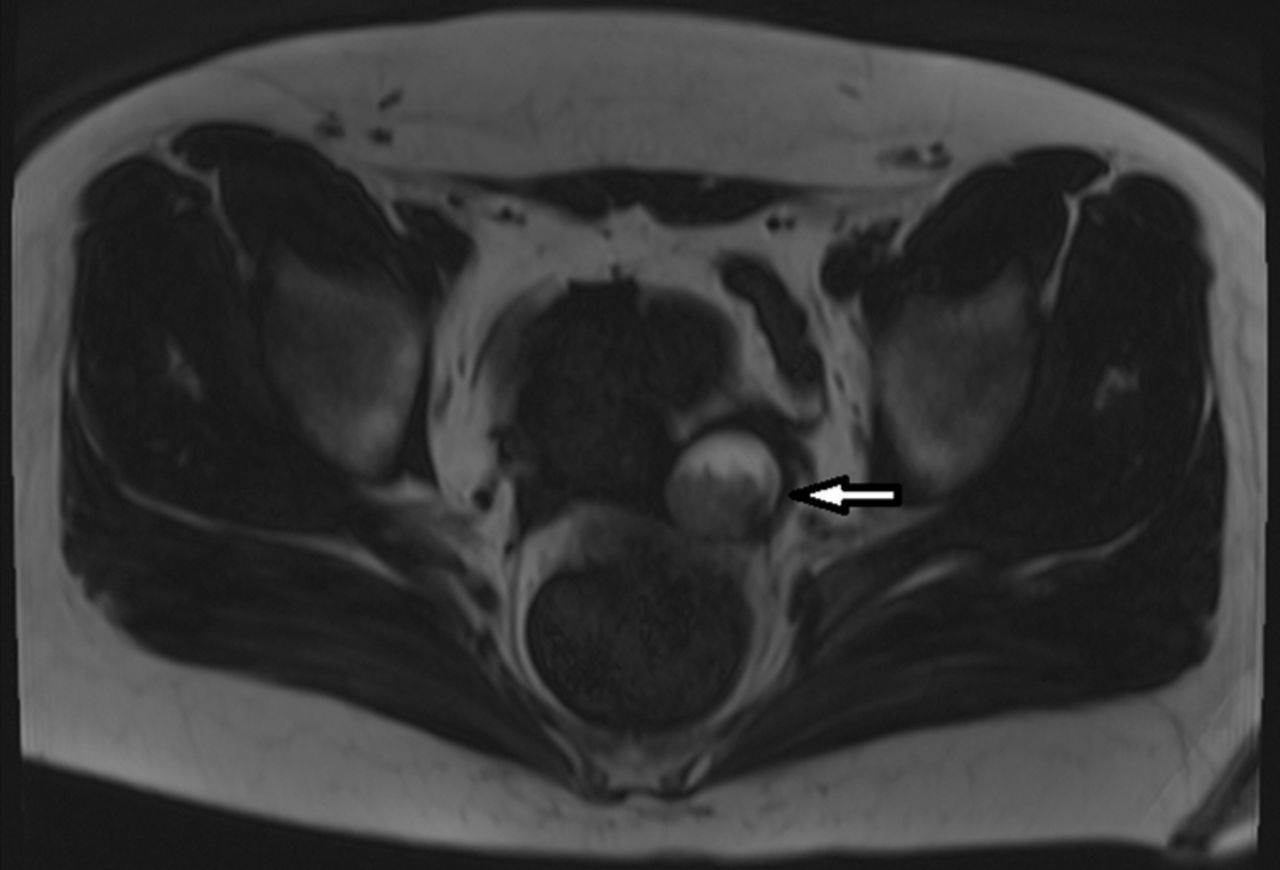
Рисунок 6. МРТ органов малого таза с рецидивом тератомы левого яичника.
Левосторонняя овариэктомия и пульс-терапия глюкокортикоидами привели к улучшению состояния и снижения выраженности симптомов. Опсоклонус и миоклонус полностью исчезли после второй операции, однако сохранялись легкая атаксия, замедленная речь и поведенческие расстройства. Через 10 месяцев из симптомов осталась только легкая атаксия.
Обсуждение
Опсоклонус-миоклонус синдром/атаксия (ОМС) является редким заболеванием; зарегистрированная заболеваемость среди педиатрических пациентов составляет от 0,18 случаев на миллион населения в Соединенном Королевстве до 0,27–0,40 случаев на миллион в Японии. Предлагаемые диагностические критерии требуют наличия по крайней мере трех из четырех следующих критериев: (1) опсоклонус, (2) миоклонус, (3) изменения поведения и/или нарушения сна, (4) нейробластома. Состояние нашей пациентки соответствовало первым трем из этих критериев. Характеристики этого синдрома совпадают с другими этиологиями острой атаксии у детей и могут представлять диагностическую проблему, особенно когда симптомы развиваются не сразу, а со временем. ОМС может быть вызван множеством причин: в первую очередь, постинфекционными или паранеопластическими синдромами. Среди детей это в 50 % случаев связано с нейробластомой, у взрослых — с МКРЛ, груди и яичников. ОМС, связанные с тератомами яичников, были зарегистрированы как среди подростков, так и среди взрослых пациентов.
Считается, что патофизиология ОМС имеет аутоиммунную природу. Хотя у многих больных и не выявляются аутоантитела, пациенты с гинекологическими злокачественными новообразованиями и ОМС могут иметь антитела к ANNA-2 (Анти-Ri). В некоторых случаях также были обнаружены антитела к NMDA-рецепторам. Аутоантитела к декарбоксилазе глутаминовой кислоты (GAD) связаны с мозжечковой атаксией, «синдромом скованного человека» и лимбическим энцефалитом. В описанном клиническом случае у пациентки выявлялся повышенный уровень анти-GAD65 в сыворотке, но не в спинномозговой жидкости, и неясно, какую роль это сыграло в развитии ее заболевания.
Исследования по терапии ОМС не обладают особой доказательной силой, но наиболее объективными подходами являются иммунотерапия и резекция опухоли. Иммунотерапевтические подходы, которые используются в педиатрической практике, включают кортикостероиды, адренокортикотропный гормон, инфузии иммуноглобулина внутривенно, плазмаферез, циклофосфамид и ритуксимаб. Несмотря на то, что в практике уже использовалось множество иммунотерапевтических методов, нет установленных руководств по каждой терапевтической стратегии. Для изучения эффективности каждого варианта лечения необходимы дополнительные исследования.
В данном случае было продемонстрировано улучшение после первоначальной резекции опухоли и повторное развитие симптомов с рецидивом опухоли. Дальнейшее лечение включало как хирургическое, так и фармакологическое вмешательство (стероиды), что привело к более быстрому и стойкому улучшению состояния. Это подчеркивает особую роль иммунотерапии и резекции опухоли в лечении ОМС. Данное заболевание может протекать монофазно или иметь рецидивирующее течение. Вероятность и тяжесть рецидивов зависят от выраженности первоначальной манифестации. При ОМС наблюдаются долгосрочные нейрокогнитивные осложнения. Хотя некоторые пациенты могут выздороветь без остаточного дефицита, остается вероятность сохранения двигательных расстройств, задержки когнитивных функций и проблем с речью через много лет после успешного лечения. Во время 4-х месячного наблюдения наша пациентка даже после значительного улучшения состояния все еще испытывала легкую атаксию, изменения в поведении и замедление речи. При последующем посещении через 10 месяцев все симптомы исчезли, за исключением очень легкой атаксии.
В педиатрической практике клиницисты обычно связывают ОМС с нейробластомой. Педиатрам следует расширить свой дифференциальный диагноз, включив в него тератому яичника, особенно среди пациентов подросткового возраста. Паранеопластический ОМС — редкий, но потенциально инвалидизирующий синдром. Важна ранняя диагностика заболевания, несмотря на потенциальные диагностические сложности. Для выработки рекомендаций по иммунотерапевтическому лечению необходимы дополнительные исследования.
Автору показалось важным описать именно анти-Hu и анти-Ma ассоциированные паранеопластические синдромы, потому что именно эти антитела могут провоцировать развитие разнообразных клинических проявлений, в случае чего может быть характерен, а в дальнейшем, возможно, и описан любой неврологический синдром.
В целом, главной задачей является заподозрить наличие паранеопластического синдрома, не упустить его клинические проявления. Учитывая, что при исследовании наличия антител будет захватываться весь спектр, дабы ничего не упустить, кратко упомянем и другие антитела (Таб. 1).
Таблица 1. Антитела, ассоциированные с поражением ЦНС при паранеопластических и некоторых аутоиммунных синдромов.
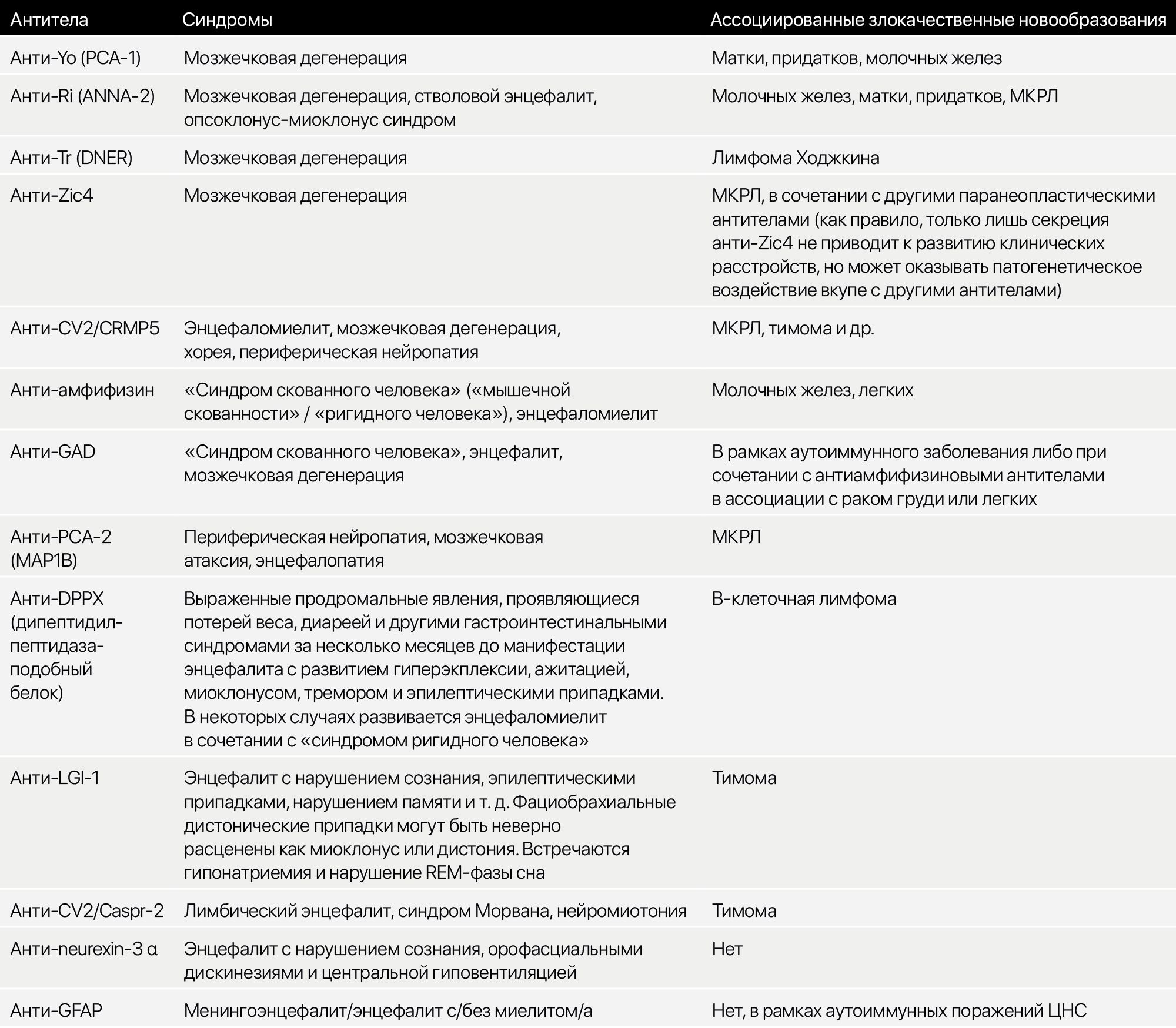
Учитывая ограниченность ресурсов, важно четко дифференцировать синдромы вне диагностики поражения ЦНС и ПНС с целью поиска признаков злокачественного новообразования или системных аутоиммунных заболеваний.
Однако, несмотря на все попытки, как уже говорилось ранее, паранеопластические синдромы очень коварны, могут манифестировать задолго до проявления злокачественного новообразования; антитела могут не обнаруживаться ни в сыворотке, ни в ликворе; при формировании антител к онконевральным антигенам (внутриклеточным) заболевание может плохо поддаваться иммунотерапии, что при его прогрессировании и невозможности обнаружения и лечения новообразования приведет к гибели пациента.
В заключении хотелось бы добавить, что паранеопластическое заболевание может развиваться в любую сторону. Принимая во внимание мутагенные процессы, возникающие в опухолевых клетках, стоит учитывать вероятность появления новых онконевральных антител у пациентов, у которых уже выявлялись другие антитела, с прогрессированием заболевания. К проявлениям, изначально рассматривавшимся в рамках хронической воспалительной демиелинизирующей полинейропатии, могут присоединиться мозжечковая атаксия и лимбический энцефалит, требующие пересмотра терапии, как это продемонстрировано в клиническом случае от Andre Dik et al. [19]
Источники:
- Titulaer M. J. et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force // European Journal of Neurology. – 2011.
- Antoine J. C., Camdessanché J. P. Paraneoplastic neuropathies //Current opinion in neurology. – 2017.
- Graus F., Dalmau J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors //Nature Reviews Clinical Oncology. – 2019.
- Kolb J. P. et al. Programmed cell death and inflammation: winter is coming //Trends in immunology. – 2017.
- Pignolet B. S. L., Gebauer C. M. T., Liblau R. S. Immunopathogenesis of paraneoplastic neurological syndromes associated with anti-Hu antibodies: a beneficial antitumor immune response going awry // Oncoimmunology. – 2013.
- Graus F. et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients // Brain. – 2001.
- Shin K. J., Ji Y. I. Anti‐Hu antibody‐mediated limbic encephalitis associated with cervical cancer: A case report //Journal of Obstetrics and Gynaecology Research. – 2018.
- Ryan T. Muir, Agessandro Abrahao, David Fam, Lorne Zinman. Clinical Reasoning: A 64-year-old woman with progressive leg weakness and ophthalmoplegia. Neurology Oct 2020, 95 (15) e2170-e2173.
- Stålberg E. et al. Standards for quantification of EMG and neurography. The International Federation of Clinical Neurophysiology //Electroencephalography and clinical neurophysiology. Supplement. – 1999.
- Li J., Lin W. Various clinical features of patients with anti-Hu associated paraneoplastic neurological syndromes: An observational study //Medicine. – 2018.
- Dimitrios D., Vaishnavi G. N., Jacob J. Paraneoplastic Confusion: A Case of Anti-Hu Encephalitis //Cureus. – 2020.
- Mehta P. R. et al. 09.12 Hu dunnit? Anti-Hu associated paraneoplastic longitudinally extensive transverse myelitis, secondary to small-cell lung cancer. – 2019.
- Pranzatelli M. R., McGee N. R. Neuroimmunology of OMS and ANNA-1/anti-Hu paraneoplastic syndromes in a child with neuroblastoma //Neurology-Neuroimmunology Neuroinflammation. – 2018.
- Hersh B. et al. Paraneoplastic opsoclonus‐myoclonus associated with anti‐Hu antibody //Neurology. – 1994.
- Bataller L. et al. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus–myoclonus //Brain. – 2001.
- Rosenfeld M. R. et al. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins //Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. – 2001.
- Amy Kunchok, M.B., B.S., and Andrew McKeon, M.D. Opsoclonus in Anti-Ma2 Brain-Stem Encephalitis. N Engl J Med 2020; 383:e84. September 24, 2020.
- Elizabeth Brigham, Chichun Sun, Richard Bronnenkant, Ashutosh Kumar, Kristin Disori. Clinical Reasoning: An adolescent girl presenting with worsening vertigo, headache, and ataxia. Neurology Sep 2020, 95 (12) e1760-e1763.
- Dik A. et al. Onconeural antigen spreading in paraneoplastic neurological disease due to small cell lung cancer //Oxford medical case reports. – 2018.
