
Сахарный диабет как нарушение всех видов обмена веществ

Глава 1 | Метаболизм азотистых оснований
Глава 2 | Биосинтез макромолекул
Глава 3 | Катаболизм и биоэнергетика
Глава 4 | Катаболизм липидов
Глава 5 | Окислительное фосфорилирование и электрон-транспортная цепь
Глава 6 | Нарушение метаболизма пуринов и пиримидинов
Сахарный диабет
Заболевание, о котором слышал даже последний троечник. Суть его в том, что имеющаяся в крови глюкоза не может проникнуть в клетки инсулинзависимых тканей (таких как жировая и мышечная). Вся беда из-за инсулина: либо его нет, либо по каким-то причинам он не действует на клетку (не секретируется должным образом; «сломались» инсулиновые рецепторы в клетке или мутировали гены протеинкиназ, ответственных за пострецепторную передачу с инсулиновых рецепторов, — да мало ли причин). Бывает так, что усиливается действие контринсулярных гормонов: глюкагона, глюкокортикостероидов (так, выделяют даже глюкокортикостероидный сахарный диабет). Бывает, что диабет становится «продолжением» иной патологии — в наихудшем случае диабет оказывается проявлением очень опасного и трудно поддающегося лечению заболевания — рака поджелудочной железы.
Классификация сахарного диабета на сегодняшний день является чрезвычайно широкой, хотя она и не претерпела существенных изменений с 1999 года (расширились лишь представления о других специфических формах сахарного диабета).

Сахарный диабет имеет множество причин, и представления об этом заболевании выходят далеко за рамки деления на диабет I и II типов. И чтобы понять, что вообще происходит при сахарном диабете, разберемся с ролью инсулина в метаболических процессах.
Механизм действия инсулина
Все, что делает инсулин с метаболизмом, направлено на выживание клетки, на ассимиляционные и пластические процессы (анаболизм). По разным данным, эффект инсулина основан на его действии на соответствующие мембранные рецепторы, обладающие тирозинкиназной активностью. Дальнейшие изменения связаны с активностью рецептора и фосфорилированием его субстратов. И здесь начинается самое интересное и самое сложное.
Внутриклеточные изменения при активации инсулинового рецептора развиваются по двум направлениям — фосфатидилинозитол-3-киназному и по МАРК-пути (см. главу 2).
События, связанные с образованием фосфатидилинозитол-3,4,5-триcфосфата, запускают в клетке метаболические изменения, которые затрагивают все виды обмена веществ. Посредником этих изменений выступает ось протеинкиназа В (АКТ) — mTOR (мишень рапамицина). Данная ось работает в нескольких направлениях:
- изменения в транскрипции ДНК и экспрессии белков, включенных в метаболические процессы;
- перемещение переносчика глюкозы GLUT 4 в клеточную мембрану;
- изменения концентрации цАМФ в клетке и модуляция активности различных ферментов в клетке.
Все это порождает метаболические ответы в клетке. Проследим же их [1, 3, 5].
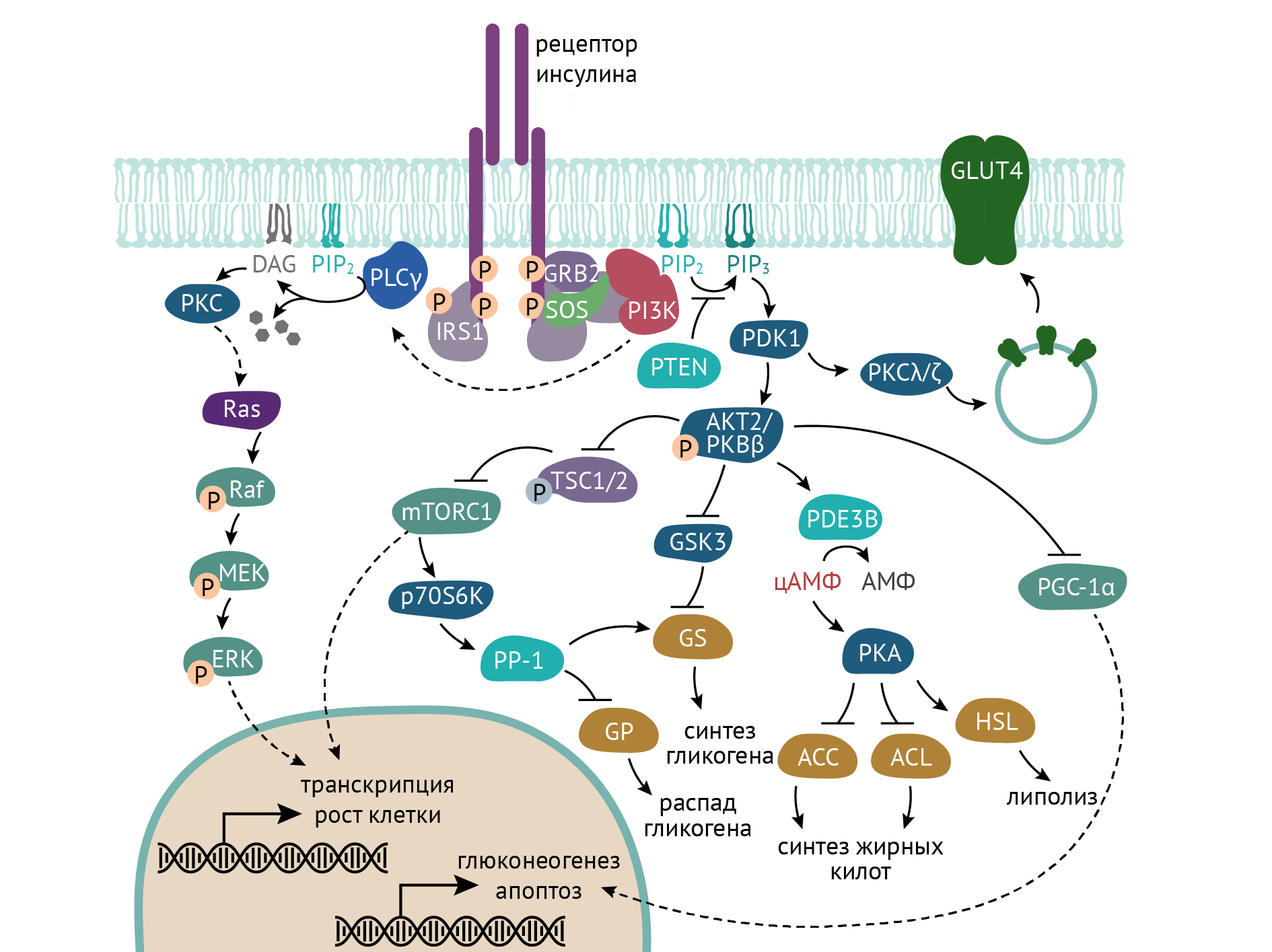
Изменения углеводного метаболизма
В целом, сводятся к повышению утилизации клетками глюкозы:
- AKT2-зависимая транслокация переносчиков глюкозы GLUT4 в плазматическую мембрану клетки. Это событие обеспечивает поступление глюкозы в инсулинзависимые ткани — мышечную и жировую.
- Торможение распада гликогена путем ингибирования фосфорилазы гликогена вследствие активации гликогенсинтазы (см. схему выше).
- AKT-зависимая активация ключевого фермента гликолиза — фосфофруктокиназы, а по ряду данных — еще и гексокиназы.
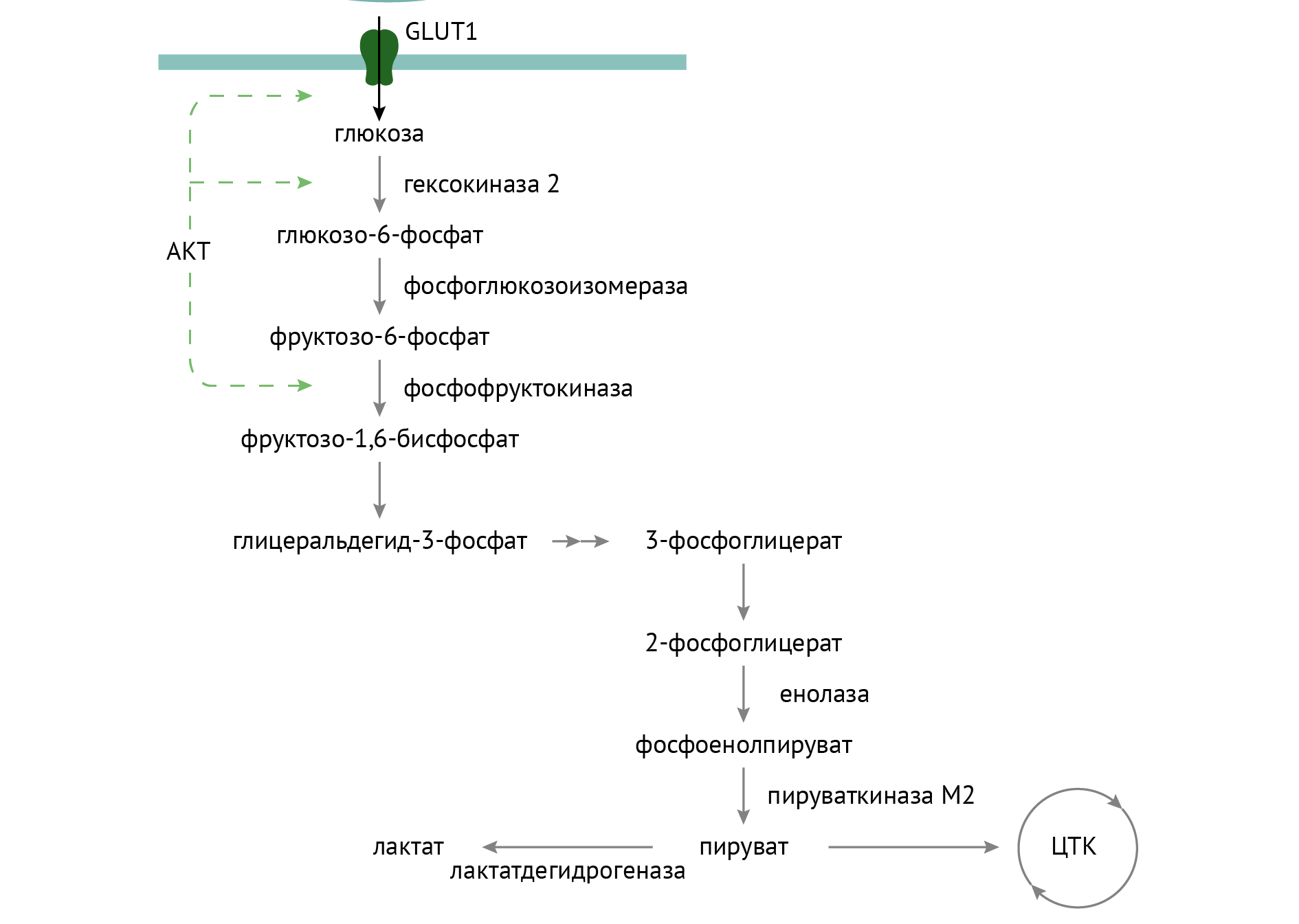
- Торможение процессов глюконеогенеза. Реализуется это как аллостерическим ингибированием фруктозо-1,6-бифосфатазы (через цАМФ-АМФ-зависимые механизмы), так и в результате изменения транскрипции генов, кодирующих ферменты глюконеогенеза. Во многом, кстати, за счет активации аденозинмонофосфат - зависимой протеинкиназы (АМФК), которая является сенсором энергодефицита - ее активность повышается как в результате воздействия на клетку инсулина, так и в результате роста АМФ в клетке (роста соотношения АМФ к АДФ, свидетельствующее об активном использовании АТФ клеткой).
Изменения липидного метаболизма сводятся к повышению запасания ТАГ [1, 3]:
- Снижение уровня цАМФ в клетке, наблюдаемое при воздействии инсулина, уменьшает активность протеинкиназы А, необходимой для активации триацилглицеридлипазы (ТАГ-липазы). Соответственно, при действии инсулина замедляется распад ТАГ и обеспечивается их сохранение в клетке.
- Активация ключевых ферментов биосинтеза длинноцепочечных жирных кислот.
Инсулин способствует катализу на важном этапе синтеза жирных кислот, и начинается это с активации ацетил-КоА-карбоксилазы.
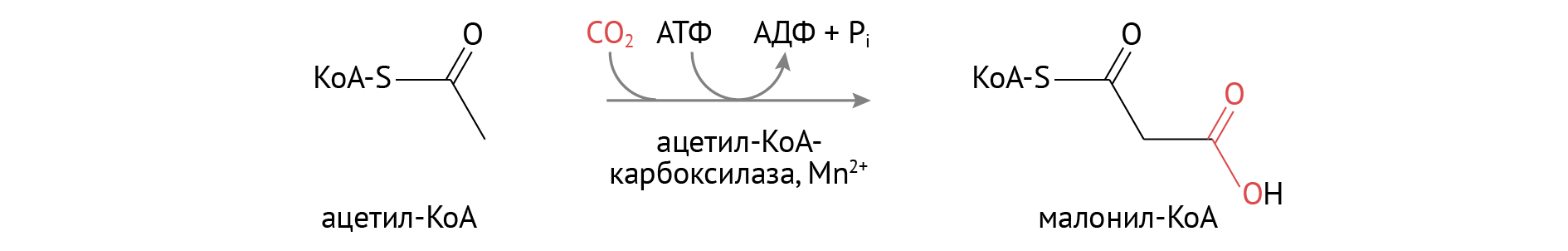
Затем малонил-КоА вовлекается в реакции биосинтеза жирных кислот, включающие в себя:
- конденсацию малых углеродных цепей;
- формирование длинной цепи, которая в конце пути станет жирной кислотой. Ниже показаны первые этапы этого во всех отношениях сложного процесса.
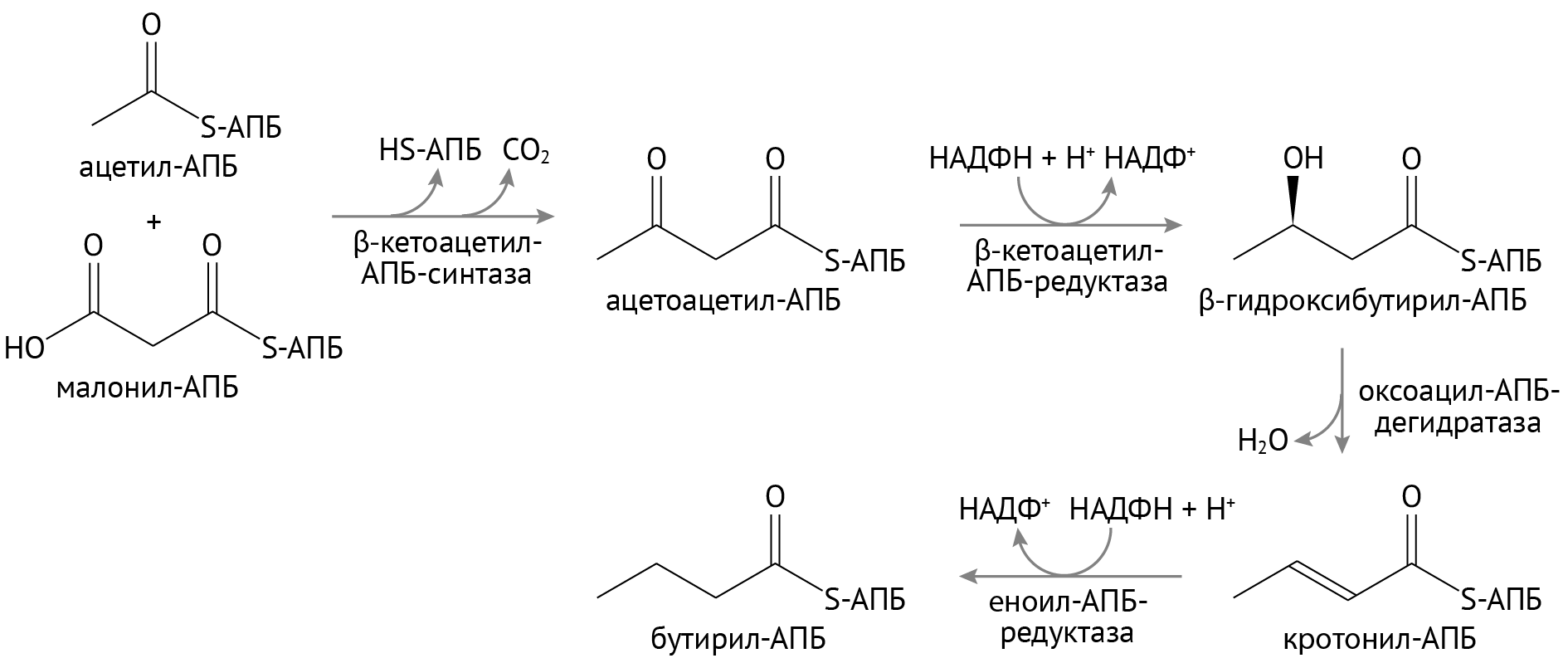
В дальнейшем образованные таким образом жирные кислоты соединяются с глицерофосфатом.
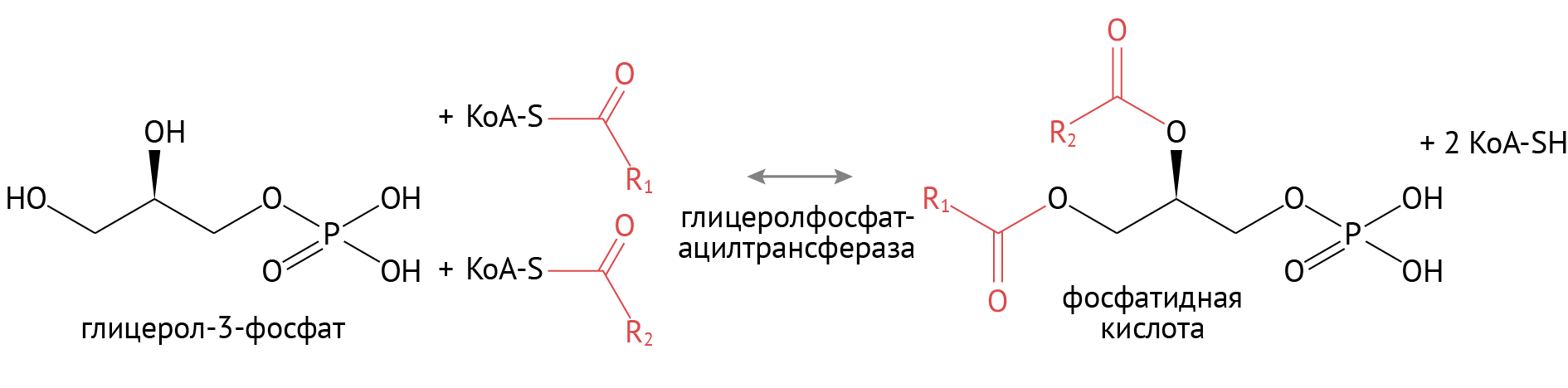
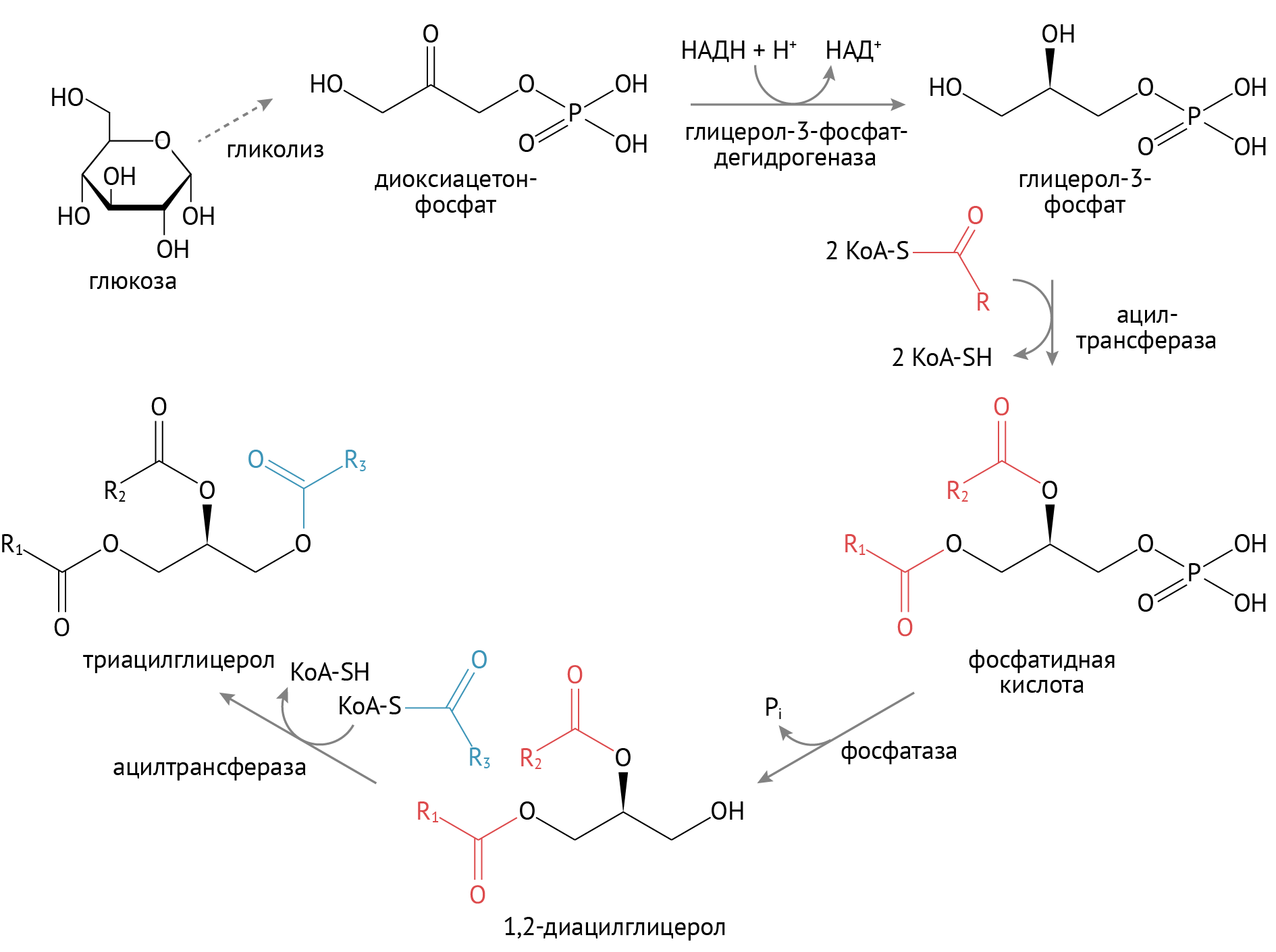
Источником глицерол-3-фосфата является уже известный тебе гликолиз. При повышенном поступлении глюкозы происходит образование множества восстановительных эквивалентов в цикле Кребса, что останавливает течение второго этапа гликолиза. Глицерол-3-фосфат в этих условиях используется для синтеза ТАГ. Именно поэтому при избыточном потреблении углеводов лишний вес — ожидаемое явление.
Действие инсулина на аминокислотный и белковый обмен [2]:
- Торможение глюконеогенеза снижает степень вовлечения аминокислот в процессы превращения их в соответствующие кетокислоты и в образование глюкозы. Это позволяет им интенсивнее участвовать в биосинтезе белка.
- По разным данным, инсулин ускоряет течение всех фаз биосинтеза белка путем активации факторов элонгации mTOR-зависимым путем:
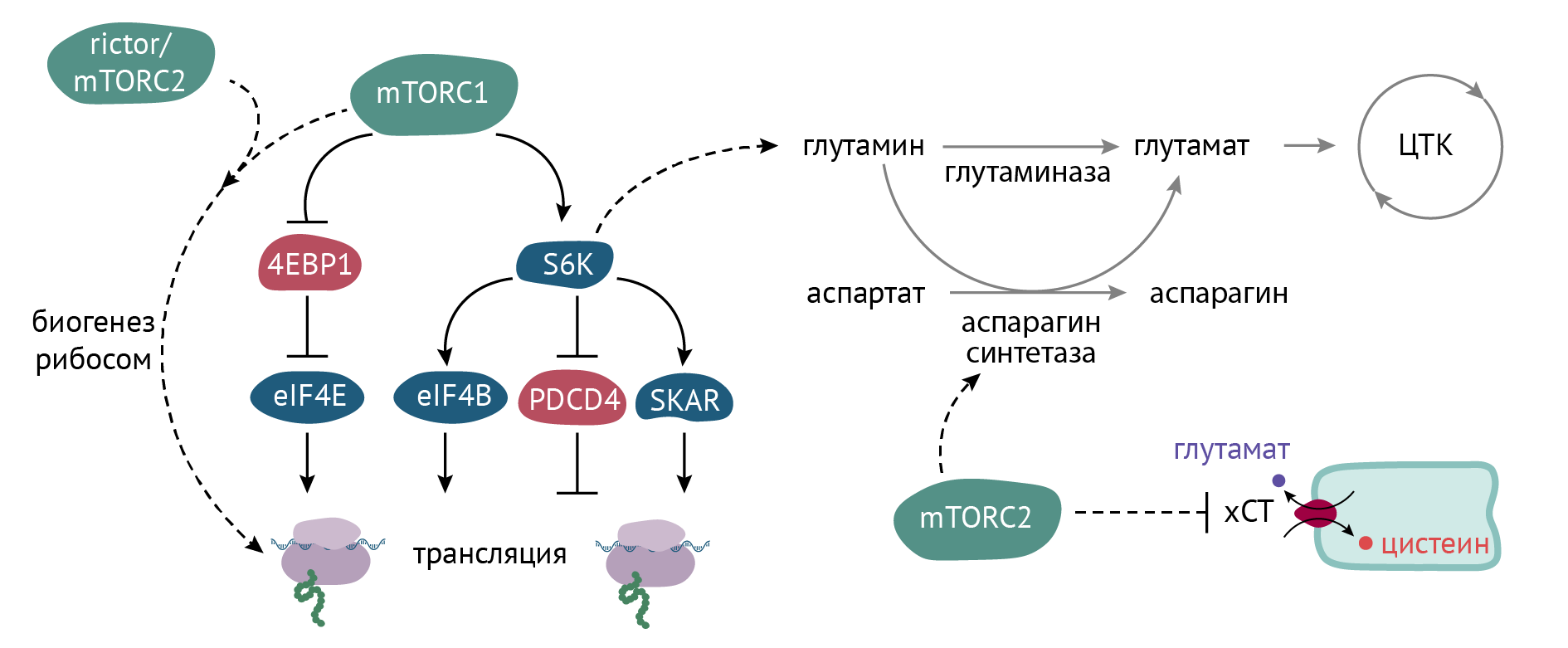
- Изменения, связанные с активацией МАРК-путей.
Отдельно стоит упомянуть и об этом моменте. На схеме выше вы могли видеть запуск митоген активного протеинкиназного каскада (SOS — RAS — RAF — MEK) от инсулинового рецептора. Данный каскад поддерживает процессы фосфорилирования внутриядерных транскрипционных факторов (ERK и прочих), что неминуемо приводит к и изменениям экспрессии генов, отвечающих за за смену фаз клеточного цикла (циклины D, E, циклин-зависимые киназы (CDK) 4, 6, 1, 2).
Они заставляют клетку переходить из фазы G1 в S. В синтетической фазе активируется крайне важный транскрипционный фактор — E2F. Он-то и опосредует запуск программы реализации синтетических процессов, необходимых для пролиферации, которые мы обсуждали во второй статье данного цикла.
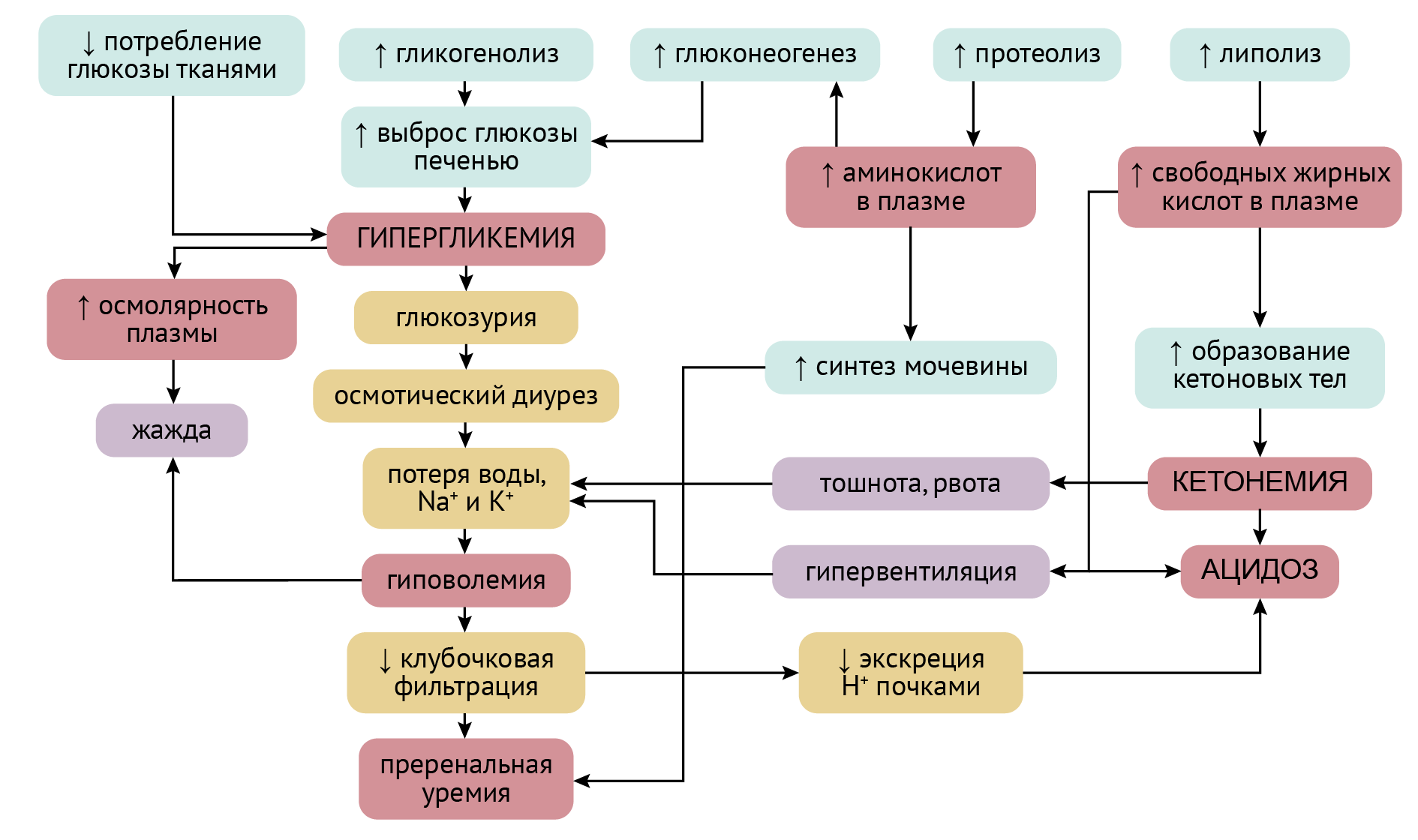
Метаболические последствия гипергликемии и отсутствия инсулина
В отсутствие инсулина в печени и почках усиливается глюконеогенез, что приводит к постоянному расходованию аминокислот и повышению уровня белкового катаболизма.
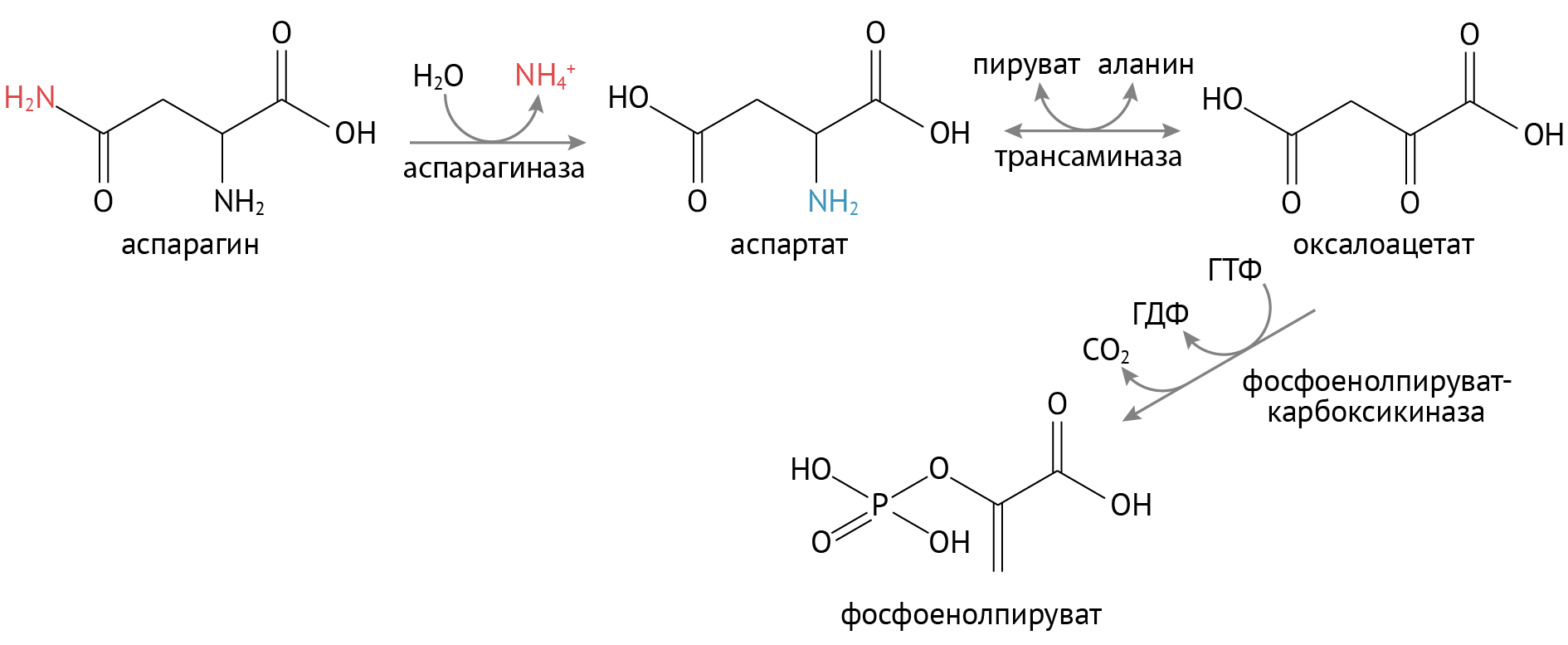
Так, аспарагин в реакции трансаминирования (АсТ) становится оксалацетатом, который превращается в фосфоенолпируват.
Снижение пула оксалацетата ослабляет течение ЦТК: ведь мы с вами помним, что уменьшение количества субстрата биохимической реакции снижает ее интенсивность. Утилизация оксалацетата в процессах глюконеогенеза улучшает биоэнергетику клетки и снижает образование АТФ. Но глюконеогенез без инсулина, несмотря на снижение уровня АТФ, лишь усиливается, и концентрация глюкозы в клетке растет.
При этом утилизировать ее все равно сложно в силу отсутствия стимулирующего влияния инсулина на фосфофруктокиназу — ключевой фермент гликолиза, катализирующий термодинамически необратимую стадию этого процесса [3, 5]. Я уж не говорю о нарушении GLUT4-зависимого транспорта глюкозы в клетку и о вовлечении аминокислот в процессы глюконеогенеза и катаболизм белков.
Второй момент: в отсутствие инсулина жиры начинают интенсивно расщепляться (организм пытается получить энергию хоть откуда-нибудь). Повышенное образование жирных кислот из ТАГ приводит к увеличению их уровня в плазме. Усиливается их β-окисление, образуется много ацетил-КоА, но вот проблема: относительно уменьшающегося содержания оксалацетата ацетил-КоА становится чрезвычайно много. Природа не терпит пустоты, и из этого избытка ацетил-КоА образуются кетоновые тела.
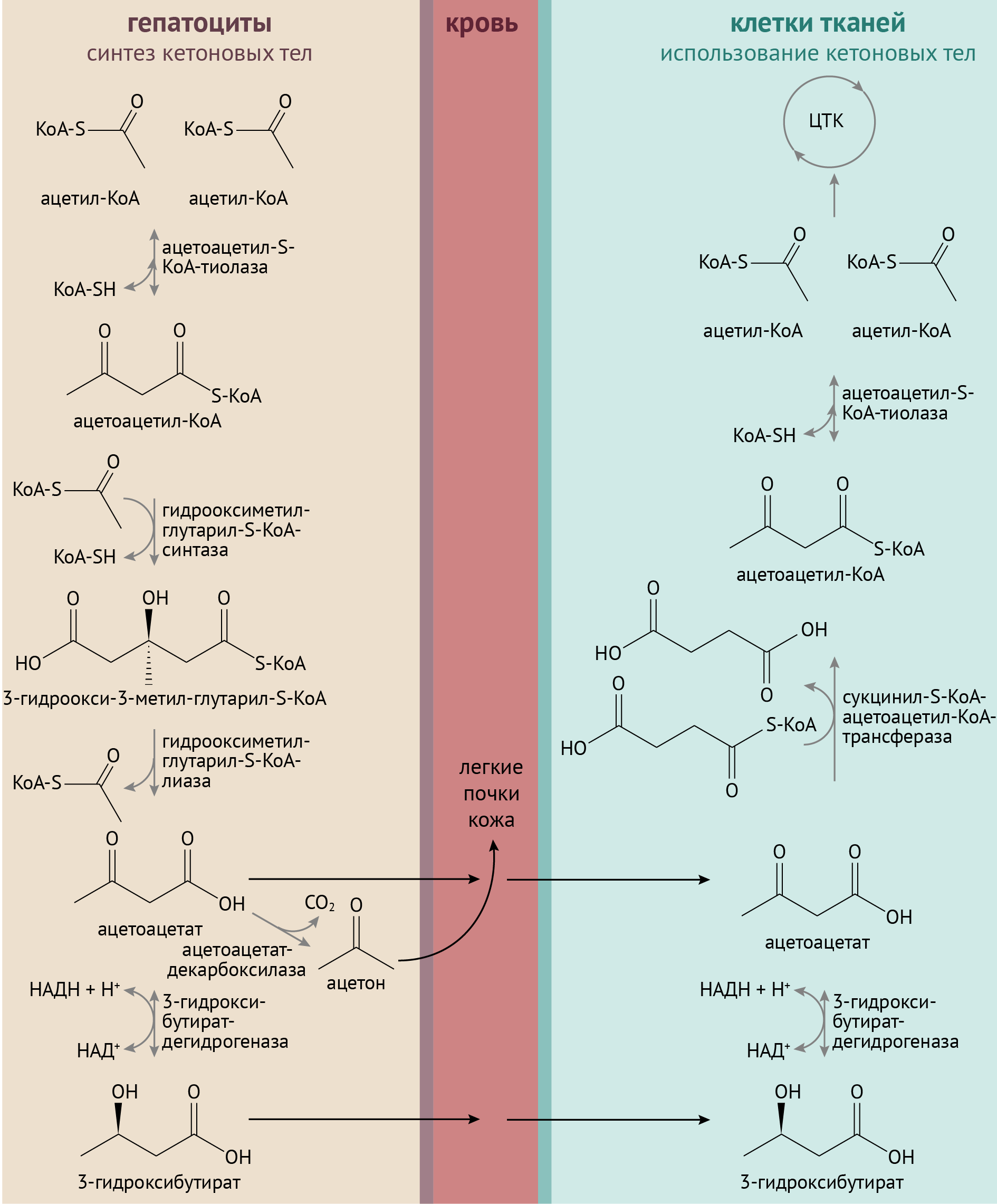
Клинические последствия метаболических изменений
Инфекционные заболевания
В условиях повышенного белкового катаболизма (во многом связанного с усилением глюконеогенеза) возникает белковая недостаточность. Страдает от этого все, особенно иммунная система. Белок необходим для синтеза антител, интерлейкинов и прочих штук, которыми располагает иммунитет, реализуя защитный ответ против чужеродных антигенов. Есть данные, что гипергликемия служит непосредственной причиной нарушения функции лейкоцитов. Все это приводит к тому, что люди с диабетом часто страдают от некоторых инфекционных заболеваний. Так, при диабете увеличивается риск возникновения инфекций желудочно - кишечного тракта (соотношение шансов наступления данного явления — 1,38 (95 ДИ 1,15–1,67,p < 0.001), кожи и мягких тканей (соотношение шансов — 1,48 (95 ДИ 1,21–1,69, p < 0,001).
Нередко у них встречается и носительство синегнойной палочки — особенно при хронических неспецифических заболеваниях легких (бронхоэктатическая болезнь). Туберкулез у таких больных — особенно его реактивация после лекарственного лечения — тоже, к сожалению, не редкость.
Метаболический ацидоз
В условиях усиленного кетогенеза возникает метаболический ацидоз, так как кетоновые тела — это существенный источник протонов, и уровень их диссоциации коррелирует со степенью закисления внутренней среды. Изменение концентрации протонов до поры до времени компенсируется работами буферных систем (бикарбонатной, белковой и прочих), но в конечном итоге этого становится недостаточно. Развивается метаболический ацидоз, имеющий решающее значение в декомпенсации сахарного диабета.
Гиповолемия, усугубление метаболического ацидоза и его влияние на функцию внутренних органов
Гипергликемия превращается в глюкозурию при преодолении порога в 9–11 ммоль/л (по разным данным). Будучи сильно водорастворимым соединением, глюкоза поступает в почки и оказывает диуретический эффект, т. к. тянет за собой воду. С мочой теряются и электролиты. Все это приводит к гиповолемии.
По последним данным, при неконтролируемом сахарном диабете парадоксальным образом почечная реабсорбция глюкозы увеличивается (имеет место дезадаптация защитного механизма, позволяющего почкам экскретировать глюкозу при повышении уровня сахара в крови), а почечный порог глюкозы смещается к более высоким значениям гликемии. Ключевую роль в этом играет повышенная активность белков-транспортеров глюкозы, преимущественно SGLT2, что способствует возрастанию реабсорбции глюкозы в почках и поддержанию хронической гипергликемии.
Гиперкалиемия
В ответ на ацидоз из клеток выходит калий, пытаясь компенсировать этот беспредел, но это у него получается плохо [7, 8].
При декомпенсации сахарного диабета в виде гипергликемии и лактатного ацидоза инсулинотерапия приводит к возвращению калия в клетку, что может стать причиной угрожающей жизни гипокалиемии. В этом случае лабораторная оценка уровня калия в крови не всегда отражает реальное его содержание в организме.
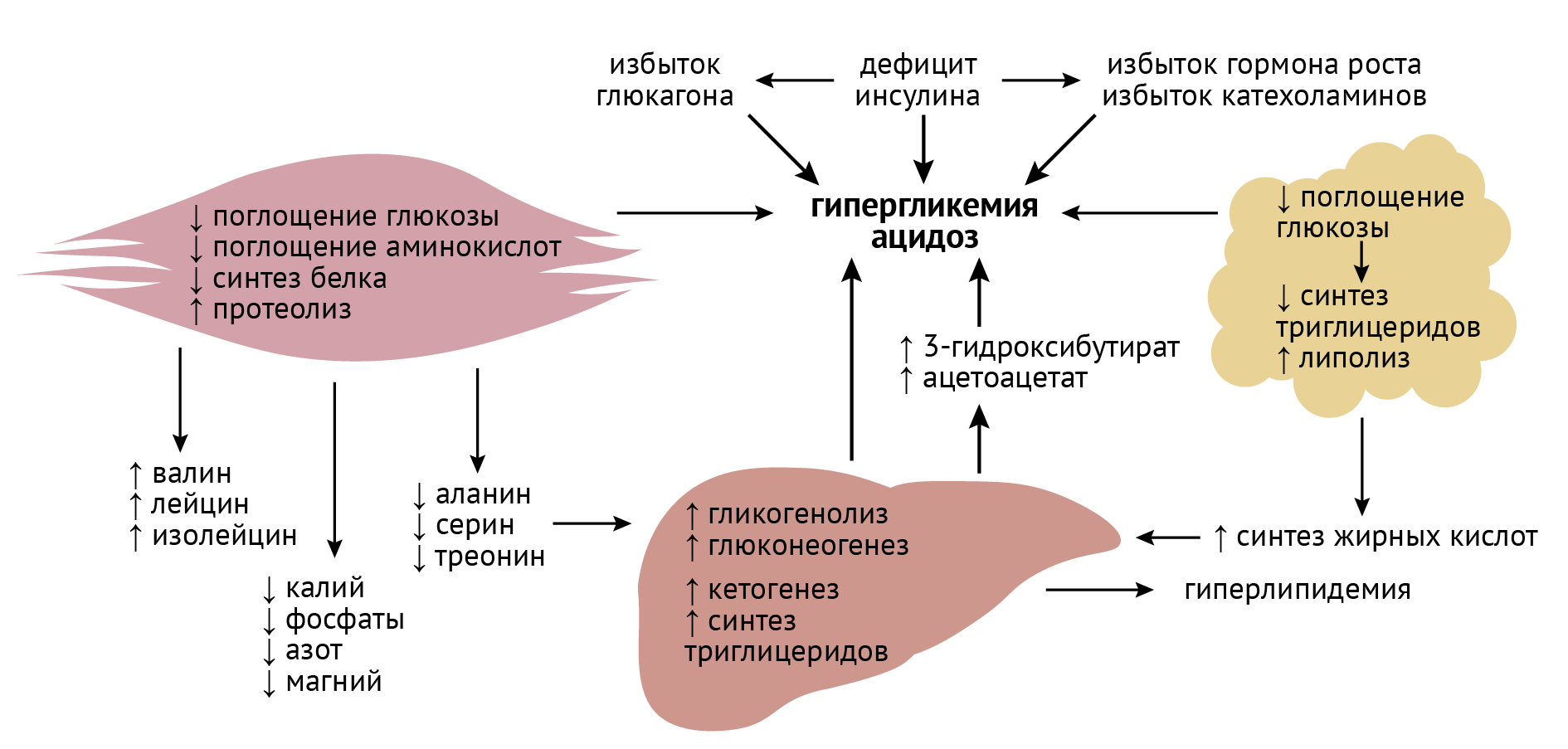
Декомпенсация и развитие полиорганной недостаточности
Стимуляция кетогенеза, гипергликемия, потеря воды и электролитов — натрия, калия, хлора, бикарбонатов (в том числе как результат ацидоза) — составляют потенциально смертельную метаболическую комбинацию. Все эти изменения чреваты развитием полиорганной недостаточности. Ведущими линиями декомпенсации будут метаболический ацидоз и циркуляторная гипоксия (связанная, главным образом, с гиповолемией).
Последняя является причиной ухудшения почечного кровотока и развития азотемии (чем и почему опасна азотемия декомпенсации, мы рассмотрим в будущих главах). Главной мишенью и решающим элементом в декомпенсации, существенно ухудшающим прогноз, будет поражение нервной системы — как в результате циркуляторной гипоксии и метаболического ацидоза, так и вследствие вторично развивающейся на этом метаболическом фоне острой преренальной почечной недостаточности.
Надо сказать, что наиболее часто декомпенсация свойственна людям с диабетом I типа: для них очень характерно развитие диабетического кетоацидоза. Для диабета II типа и иных, специфических типов диабета развитие кетоацидоза является редкостью (но «редко» — не значит «никогда»).
Длительное течение сахарного диабета приводит к поражению сосудов среднего и мелкого калибра, которое носит название диабетической ангиопатии. В целом, это выражается в реализации дистрофических и фиброзно-пролиферативных процессов в стенках сосудов, что существенно ухудшает микроциркуляцию и транскапиллярный обмен между сосудами и тканями, а также кровоснабжение сосудистых стенок. Наиболее выраженные изменения возникают в почках (диабетическая нефропатия и нефросклероз — одна из ведущих причин хронической почечной недостаточности), в сетчатке глаза (диабетическая ретинопатия — постепенное ухудшение зрения вплоть до слепоты), сосудах, питающих нервные волокна (нейропатия, развитие вегетативных нарушений — нарушение функции тазовых органов; развитие двигательных и чувствительных неврологических нарушений; в ряде случаев это проявляется усугублением течения ишемической болезни сердца за счет безболевой ишемии миокарда и бессимптомного течения инфаркта миокарда).
Сахарный диабет имеет системные проявления, и это особенно заметно при его комбинации с другими заболеваниями. В ряде случаев диабет играет роковую роль, утяжеляя их течение. В особенности это касается атеросклероза и гипертонической болезни, что связано с уже упомянутой диабетической ангиопатией. При ее наличии атеросклероз становится более распространенным и затрагивает сосуды мышечного типа среднего калибра, что не характерно для такого заболевания при отсутствии сахарного диабета.
О роли сахарного диабета в течении других болезней и его последствиях можно говорить бесконечно. Особого внимания заслуживает повышенный риск возникновения и усугубление течения инфекционных заболеваний в целом, и туберкулеза, синегнойной инфекции, кандидоза и других видов микозов — в частности. Больные диабетом требуют особого подхода и в хирургической практике. Операция является для организма стрессовой ситуацией, на которую он отвечает неспецифично — выбросом глюкокортикостероидов и активацией симпато-адреналовой системы. Как ты уже понял, в этих условиях возникает гипергликемия, в силу чего такие пациенты нуждаются в консультации с эндокринологом; подробнее об этом рассказано тут.
Многие заболевания у пожилых пациентов зачастую проявляются атипично, иначе говоря — «смазанно», без выраженной характерной для конкретного заболевания симптоматики. Например, одним из немногих симптомов пневомнии, инфаркта миокарда, острых гнойных заболеваний брюшной полости и т. д. может быть гипергликемия (т. е. повышение уровня сахара относительно «привычного» для конкретного пациента уровня) и декомпенсация сахарного диабета — опять же, в связи с неспецифическим ответом организма на возникающие повреждения в органах и тканях. Будь внимательнее.
Некоторые аспекты лекарственной терапии сахарного диабета
Дисклеймер: я далеко не эндокринолог, и представленная информация ниже скорее ознакомительная (и подразумевает дальнейшее самостоятельное погружение в проблему).
Ниже представлены некоторые группы сахароснижающих лекарств — тех препаратов, что нашли применение в терапии инсулинрезистентного сахарного диабета. Их использование направлено на увеличение эффективности воздействия инсулина на ткани. Важно помнить, что образование инсулина при диабете II типа и других вариантах, не относящихся к инсулинозависимому сахарному диабету, не прекращается, а поначалу даже является повышенным. Препараты могут влиять на самые разные уровни реализации эффектов инсулина — от его секреции из β-клеток островков Лангерганса до воздействия на клетки-мишени. Приведу лишь некоторые данные, уместные для освещения в рамках биохимии.
Производные сульфонилмочевины (глибенкламид, гликлазид, глимеприд) ингибируют калиевые каналы в β-клетках поджелудочной железы, приводя к деполяризации их плазматических мембран. Это вызывает открытие кальциевых каналов в мембранах депо кальция, что приводит к повышению внутриклеточной концентрации кальция. В результате происходит экзоцитоз инсулина.
Между нами говоря, во всем мире от них постепенно отходят в пользу более удобных и эффективных препаратов, но встретить пациентов, получающих сульфонилмочевину, по-прежнему возможно.
Инкретиномиметики (ингибиторы дипептидазы и глюкагоноподобный пептид — эксенатид, ситаглиптин, вилдаглиптин, саксаглиптин) осуществляют активацию соответствующих рецепторов на мембране β-клеток; последующий рост уровня цАМФ приводит к усилению секреции инсулина.
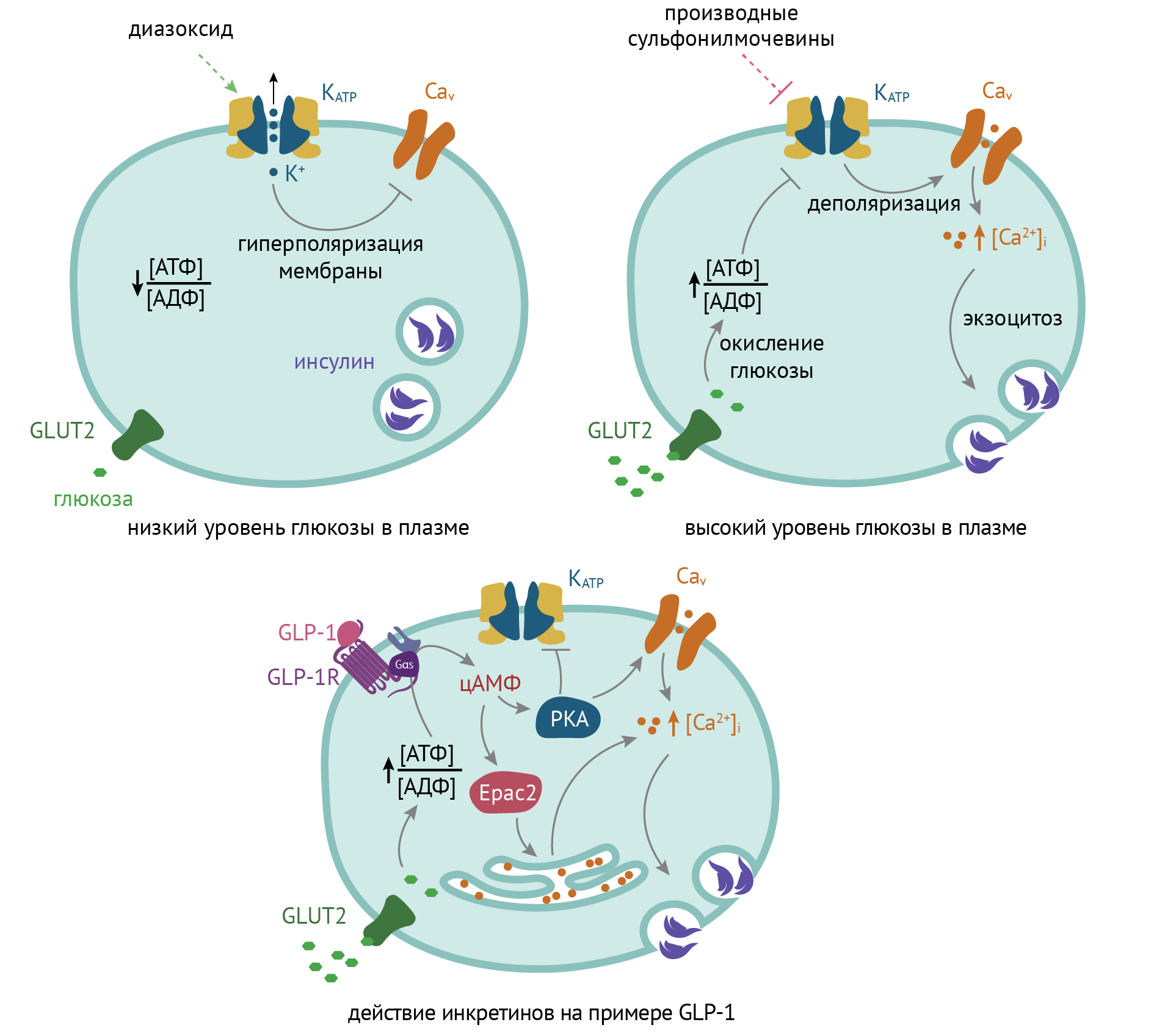
(Напоминаю о том, что К+-каналы в β-клетках могут ингибироваться при повышении уровня цАМФ, которое происходит под действием инкретинов.)
Бигуаниды (метформин) способствуют активации АМФ-зависимой протеинкиназы (АМФК). АМФ-зависимая протеинкиназа активирует транскрипционные факторы, которые подавляют экспрессию генов, кодирующих ферменты глюконеогенеза — фосфоенолпируваткарбоксикиназы и глюкозо-6-фосфатазы. С АМФК связывают и усиление транслокации GLUТ4 — переносчика глюкозы в плазматическую мембрану клеток, ведущую к утилизации клеткой глюкозы.
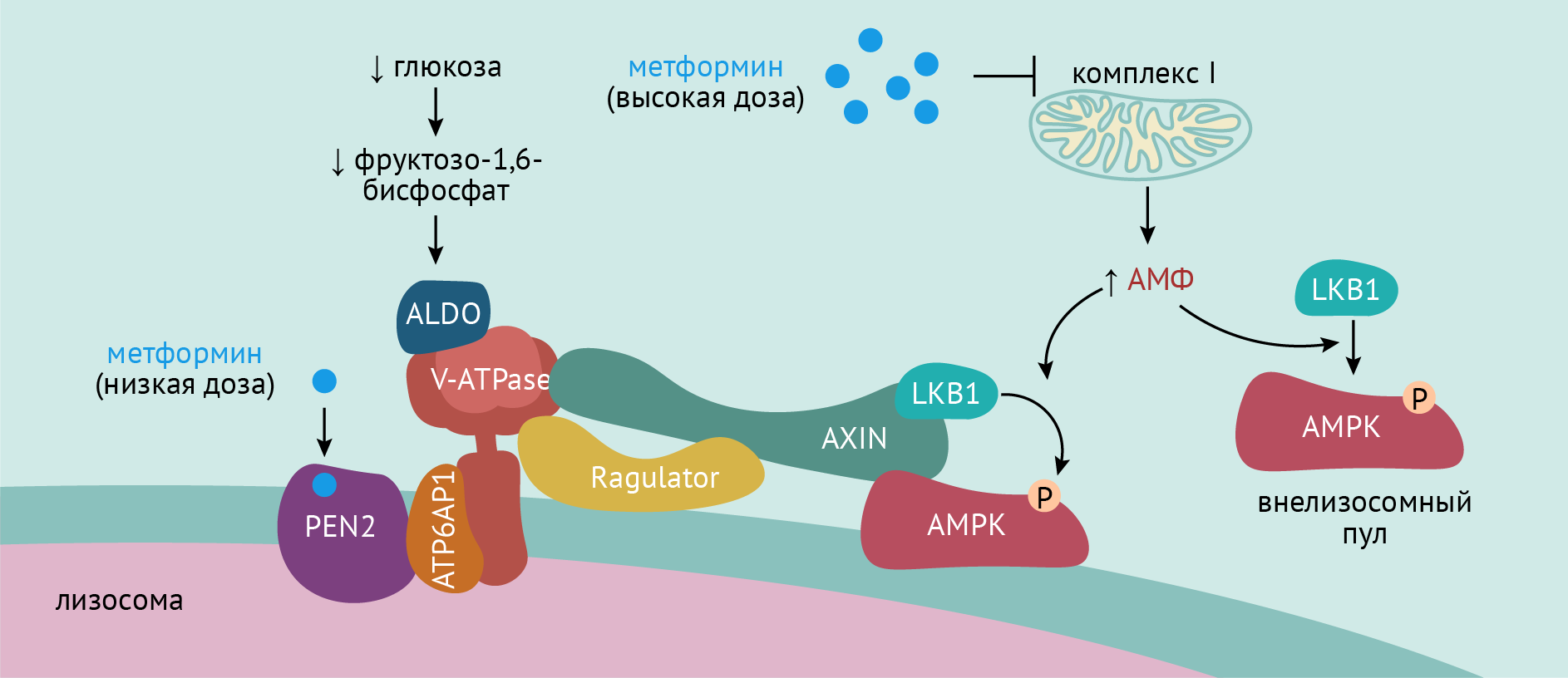
Долгое время считалось, что механизм действия бигуанидов сопряжен, главным образом, с повышением в клетках содержания АМФ (что должно повлечь за собой активацию АМФК). Однако, это реализуется лишь при высоких дозах метформина (порядка 1,5–2 г/сут), что в реальной жизни не увидишь.
Между тем, недавно миру стало известно, что активация AMФK при воздействии метформина в терапевтических дозах не сопряжена с увеличения соотношения аденозинмонофосфата/аденозиндифосфата (АМФ/АДФ), и что механизм оказывается более вычурным и сложным.
Метформина связывается с белком PEN2 (presenilin enhancer 2). PEN2 связывается с субъединицей ATP6AP1, входящей в состав АТФ-азы V (вакуолярного) типа. Сложным путем это приводит к инактивации АТФ-азы V и активации АМФК, Последствия же, в виде “имитирования” внутриклеточных эффектов инсулина на углеводный обмен, вы знаете из прочитанного ранее в этой главе. [1-3]
Глифлозины (канаглифлозин, дапаглифлозин и пр.) останавливают реабсорбцию глюкозы в почках путем ингибирования SGLT2-транспортера, что усиливает выведение глюкозы с мочой и снижает выраженность гипергликемии. Эти препараты новые, на них возлагаются большие надежды. Окончательно в клиническую практику они пока вошли недостаточно широко, но это вопрос времени [10].
- Ma, T., Tian, X., Zhang, B. et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 603, 159–165 (2022).
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014
- Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002
- Chávez-Reyes J, Escárcega-González CE, Chavira-Suárez E, León-Buitimea A, Vázquez-León P, Morones-Ramírez JR, Villalón CM, Quintanar-Stephano A, Marichal-Cancino BA. Susceptibility for Some Infectious Diseases in Patients With Diabetes: The Key Role of Glycemia. Front Public Health. 2021
- Chávez-Reyes J, Escárcega-González CE, Chavira-Suárez E, León-Buitimea A, Vázquez-León P, Morones-Ramírez JR, Villalón CM, Quintanar-Stephano A, Marichal-Cancino BA. Susceptibility for Some Infectious Diseases in Patients With Diabetes: The Key Role of Glycemia. Front Public Health. 2021
- Solá E, Rivera C, Mangual M, Martinez J, Rivera K, Fernandez R. Diabetes mellitus: an important risk factor for reactivation of tuberculosis. Endocrinol Diabetes Metab Case Rep. 2016
- Aronson, D., Rayfield, E.J. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol 1, 1 (2002)
- Sivanmaliappan TS, Sevanan M. Antimicrobial Susceptibility Patterns of Pseudomonas aeruginosa from Diabetes Patients with Foot Ulcers. Int J Microbiol. 2011;2011:605195. doi:10.1155/2011/605195
- Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007
- Magaway C, Kim E, Jacinto E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells. 2019;8(12):1584. Published 2019 Dec 6. doi:10.3390/cells8121584
- Abu-Ashour W, Twells LK, Valcour JE, Gamble JM. Diabetes and the occurrence of infection in primary care: a matched cohort study. BMC Infect Dis. 2018
- Таганович А. Д. , Олецкий Э. И., Котович И. Л.. Патологическая биохимия, 2015 г. Главы 1, 2.
- Портал «Биохимия для студента». Раздел: «Сахарный диабет».
- Тимин О. А. Основы биологической химии, 2018 г., с. 291.
- Виноградов В. М., Каткова Е. Б. Информация про противодиабетические препараты. Фармакология с рецептурой, 2016 г.
- Кольман Я. , Рём К.-Г. Наглядная биохимия, 5-е издание, 2018 г., с. 152, 382–386.
- Танбаева Г. З., «Кардиоваскулярная безопасность менеджмента сахарного диабета», МВА, 2018 г.
- Шанин В. Ю. Патофизиология критических состояний. 2-е издание, 2018 г., с. 274–281.
- Зилбернагль, Ланг Ф. Клиническая патофизиология, 2-е издание, 2016 г., с.310.
