
Нарушение метаболизма пуринов и пиримидинов

Глава 1 | Метаболизм азотистых оснований
Глава 2 | Биосинтез макромолекул
Глава 3 | Катаболизм и биоэнергетика
Глава 4 | Катаболизм липидов
Глава 5 | Окислительное фосфорилирование и электрон-транспортная цепь
Подагра и гиперурикемия
В чем отличие подагры от простой гиперурикемии? Гиперурикемия — это лабораторный феномен, суть которого — увеличение содержания в крови мочевой кислоты. Не во всех случаях гиперурикемия приводит к возникновению клинически значимых симптомов. Подагра же — состояние, характеризующееся отложением кристаллов мочевой кислоты в тканях. Больше всего достается почкам и суставам. Подагры не бывает без гиперурикемии. Но как же последняя приводит к такому тяжелому заболеванию?
Мочевая кислота имеет свой предел растворимости. В чрезмерно кислой среде (c pH ниже 5,7) она выпадает в виде кристаллов уратных солей в мягких тканях. Оседая в суставах, соли мочевой кислоты вызывают чрезвычайно болезненные ощущения. Первым достается пальцам ног. Ураты для тканей — чужеродные соединения. В силу этого в тканях реализуется типовой патологический процесс — воспаление, которое в данном случае имеет скорее разрушающее, нежели защитное действие [1].
Но почему так происходит? Бывают случаи, когда мочевой кислоты образуется слишком много.
А) Повышенное образование пуринов и их усиленный распад (логичное следствие). Здесь причиной служит генетически детерминированная чрезмерная активность фосфорибозилпирофосфаттрансферазы (катализатор первой реакции синтеза пуринов), которая становится менее чувствительной к отрицательным регуляторным факторам — ГМФ и АМФ [6].
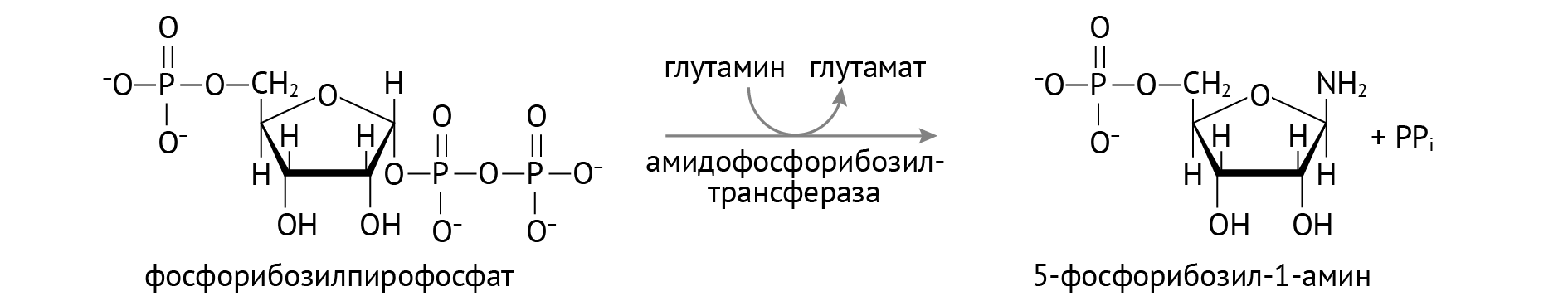
Б) Недостаточная реутилизация. Бывает так, что распадающиеся пурины утилизируются хуже, чем положено (не по плану) в силу дефектов гипоксантингуанинфосфорибозилтрансферазы (фермента, переводящего гипоксантин в ИМФ, а гуанин — в ГМФ). В результате мы получаем большое количество мочевой кислоты.
Ситуация может усугубляться, если с пищей поступает много нуклеиновых кислот, которые, распадаясь на пурины, превращаются в энтероцитах в мочевую кислоту и в таком виде всасываются в кровь. Алкоголь нарушает выведение мочевой кислоты (по пока неясным для меня самого причинам). При гиперурикемии противопоказаны диуретики (препараты, усиливающие выведение мочи, в особенности — из тиазидной группы), так как они повышают риск уратной нефропатии. Как видишь, гиперурикемия создает сплошные трудности. В отношении подагры надо сказать, что это инвалидизирующее заболевание, снижающее качество жизни и увеличивающее риск сердечно-сосудистых патологий [1].
Терапевтический подход
По вышеуказанным причинам первым вектором лечения подагры будет соблюдение диеты, которая состоит в отказе от потребления тех продуктов, которые в большом количестве содержат пуриновые основания. Исключаются пиво, кофе, чай, шоколад, мясные продукты, печень, красное вино. Предпочтение отдается вегетарианской диете с количеством чистой воды не менее двух литров в сутки.
Медикаментозный подход заключается в применении аллопуринола, конкурентного ингибитора ксантиноксидазы, — той самой, что переводит ксантины в мочевую кислоту. Это терапия первой линии [4–6].
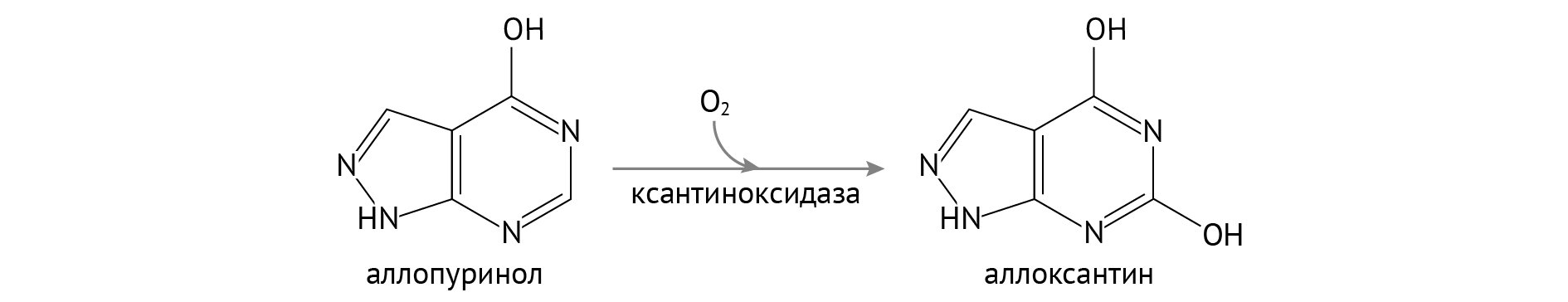
В силу того, что аллопуринол отнимает все внимание ксантиноксидазы, превращаясь в хорошо растворимый аллоксантин, ксантин и гипоксантин, будучи обиженными на сложившуюся ситуацию, покидают организм с мочой [6].
Уратная нефропатия
Уратная нефропатия создает массу проблем. Надо сказать, что сам термин «уратная нефропатия» сегодня используется реже, чем ранее, а кем-то и вовсе считается неправильным. Несмотря на это, именно так мы назовем поражение почек при гиперурикемии. Часто именно ураты формируют мочевые камни, становясь основой для мочекаменной болезни и вторичного пиелонефрита. У примерно половины людей, страдающих подагрой, присутствует еще и мочекаменная болезнь [4].
Механизм камнеобразования в данном случае провоцируется повышением кислотности (понижением рН) мочи. Это делает возможным переход мочевой кислоты в плохо растворимую форму:
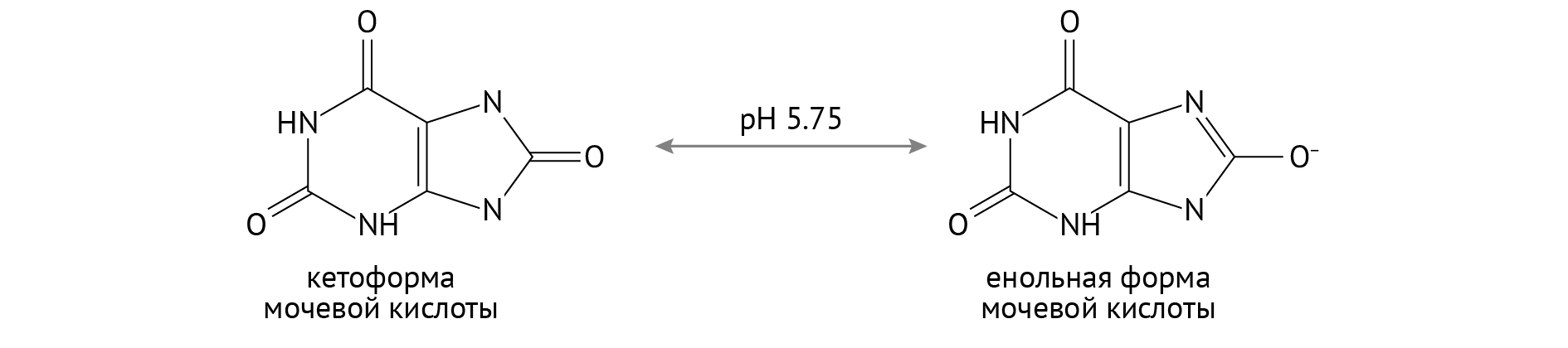
Мочекаменная болезнь опасна вследствие риска возникновения острой задержки мочи, которая становится причиной острой постренальной почечной недостаточности, а та, в свою очередь, является угрожающим жизни патологическим состоянием.
Синдром Леша — Нихана
В случае наличия мутации гена, кодирующего гипоксантингуанинфосфорибозилтрансферазу, развивается синдром Леша — Нихана. Наследуется он Х-сцепленно, а это значит, что достается практически только парням. Но не часто (реже, чем могло бы быть, но чаще, чем хотелось бы): патология грозит одному человеку из 380 тысяч.
Особенность этого синдрома в том, что здесь имеется грозная неврологическая и психическая симптоматика с самого раннего возраста: умственная отсталость и снижение темпов психомоторного развития; нарушения двигательных функций по типу хореатетоза, аутоагрессивное поведение (ребенок пытается нанести себе вред, что в условиях умственной отсталости закономерно; но по той же причине такие попытки не всегда удачны). К сожалению, патофизиологические механизмы развития поражения ЦНС при болезни Леша — Нихена не ясны по сей день. «Так, несмотря на широкий спектр проблем, связанных с гиперпродукцией мочевой кислоты, причинно-следственная связь между ними и неврологическими и поведенческими проявлениями не установлена, и терапия аллопуринолом, проводимая с момента рождения, не влияет на поражение ЦНС» [7].
Помимо поражения нервной системы такие дети не лишены и уратной нефропатии, а также подагры. Оба этих патологических состояния развиваются по рассмотренным нами выше механизмам [6, 7].
Последнее, что здесь стоит отметить: спектр клинических проявлений варьирует в зависимости от степени утраты активности фермента. Так, при активности фермента ниже 1,5 % от необходимой развивается полный набор вышеописанных клинических симптомов, в том числе и аутоагрессивное поведение, и когнитивная недостаточность. Это — следствие поражения коры головного мозга. При сохранении активности фермента в интервале от 1,5 до 8 % от необходимой признаки поражения коры, чаще всего, не прослеживаются.
Но имеются иные неврологические нарушения, проявляющиеся в разной степени выраженности экстрапирамидной и пирамидной моторной дисфункции и связанные с гиперурикемией. Сохранение активности фермента свыше 8 % сопровождается лишь уратной нефропатией (нефролитиазом — образованием камней в мочевыводящих путях) и подагрой [4, 6, 7].
Синдром лизиса опухоли
Ряд опухолевых заболеваний (чаще это гемобластозы — лейкозы, лимфомы, реже — солидные опухоли: рак желудка, легкого, молочной железы и т. д.) может давать картину острой почечной недостаточности в ответ на проводимую химиотерапию. Распадаясь и разрушаясь от действия химиотерапевтических препаратов (флударабина, метотрексата, да и многих других), опухолевые клетки выбрасывают в кровь большое количество мочевой кислоты, калия и фосфора. Последствия бывают достаточно серьезные:
- гиперурикемия провоцирует развитие нефролитиаза почечных канальцев и почечной недостаточности;
- гиперкалиемия способсвует возникновению тяжелых сердечных аритмий, вплоть до остановки сердца (усугубляется вторичной гипокальциемией) в связи с угнетением атриовентрикулярной проводимости в сердце;
- гиперлактатемия (связана с высокой активностью в опухолевых клетках анаэробного гликолиза, обусловливающего высокое содержание в них лактата) с риском развития метаболического ацидоза.
К метаболическим изменениям при синдроме лизиса опухоли также относят повышение уровня остаточного азота в крови (в частности, мочевины, которое выступает как проявление повышенного катаболизма белков клеток), вторичную гипокальциемию на фоне гиперфосфатемии, изменение кислотности крови — ацидоз.
Гиперурикемия при несоответствии возможностям почек провоцирует мочекислый нефролитиаз с развитием мочекислой нефропатии (по указанному в 1.6.2 механизму) и острой почечной недостаточности.
В настоящее время при проведении химиотерапии осуществляется профилактика синдрома лизиса опухоли [3], которая отчасти достигается высокой инфузионной (гидратационной) поддержкой таких пациентов и соблюдением режимов химиотерапии и скорости введения препаратов. Есть сообщения об успешном профилактическом применении аллопуринола у таких пациентов. Но, несмотря на это, риск развития лизиса опухоли и последующих тяжелых метаболических изменений нельзя не учитывать, когда мы имеем дело с химиотерапией злокачественных новообразований, и риск этот тем больше, чем интенсивнее режим химиотерапии. Лечение пациентов осуществляется в условиях реанимации и состоит в интенсивной инфузионной поддержке, иногда применяются попытки гемодиализа (однако есть данные об его неэффективности; более подробно смотри в специализированной литературе по онкологии и интенсивной терапии).
Тяжелый комбинированный иммунодефицит
Незаслуженно забываемый многими фермент аденозиндезаминаза может быть причиной тяжелого комбинированного иммунодефицита у детей раннего возраста. Заболевание развивается в случае мутации гена, кодирующего данный фермент. Аденозидезаминаза (АДА) превращает аденозин в инозин, тем самым участвуя в катаболизме адениловых нуклеотидов. При отсутствии АДА в клетках накапливается аденозин и дезоксиаденозин. Повышенное содержание этих метаболитов угнетает рибонуклеотидредуктазу — ту самую, что отвечает за синтез нуклеиновых кислот. Без них невозможен синтез ДНК, а значит, угнетается пролиферация клеток. Первыми отхватывают клетки крови (быстро пролиферирующие), в особенности лимфоциты, как В, так и Т. В результате развивается комбинированный иммунодефицит, т. е. дефект клеточного и гуморального адаптивного иммунитета. Все это проявляется чрезвычайно частым развитием гнойных тяжелых инфекций, которые для обычных детей не совсем характерны и приобретают рецидивирующий характер, не склонный к разрешению. Происходить это все начинает (приблизительно) с шести месяцев, когда заканчивается действие материнских антител, получаемых младенцем при грудном вскармливании. Дети болеют тяжело, часто, и причем такими инфекциями, которыми иммунокомпетентный ребенок болеть не будет (эти инфекции называют оппортунистическими) [2].
В настоящее время есть данные об успешной генотерапии (введении во взятые от пациента стволовые клетки костного мозга генетического вектора, кодирующего дезаминазу) с последующим возвращением и серией ТГСК (процедур трансплантации гемопоэтических стволовых клеток). Это два наиболее адекватных варианта лечения таких заболеваний, но, как ты понимаешь, они не везде доступны.
Не могу не отметить, что ТКИД лишь в 50 % случаев вызывается мутацией в гене, кодирующем дезаминазу. ТКИД как группа заболеваний обусловлен нарушением процесса созревания или пролиферации лимфоцитов. Существует 15 генетических вариантов данного заболевания. Иногда виноват дефект другого фермента катаболизма пуринов — пуриннуклеозидфосфорилазы, с которой мы встречались ранее.
Также причина ТКИД может быть результатом дефекта IL2RG — общей субъединицы для многих рецепторов к цитокинам (ИЛ-2/4/7/9/15/21), запускающим клеточный ответ в лимфобластах в виде их пролиферации и созревания. Бывает изолированный дефект рецептора к ИЛ-7. А может быть дефект киназы JAK3 (янус-тирозинкиназы), которая является внутриклеточным посредником сигнала, получаемого вышеуказанными рецепторами.
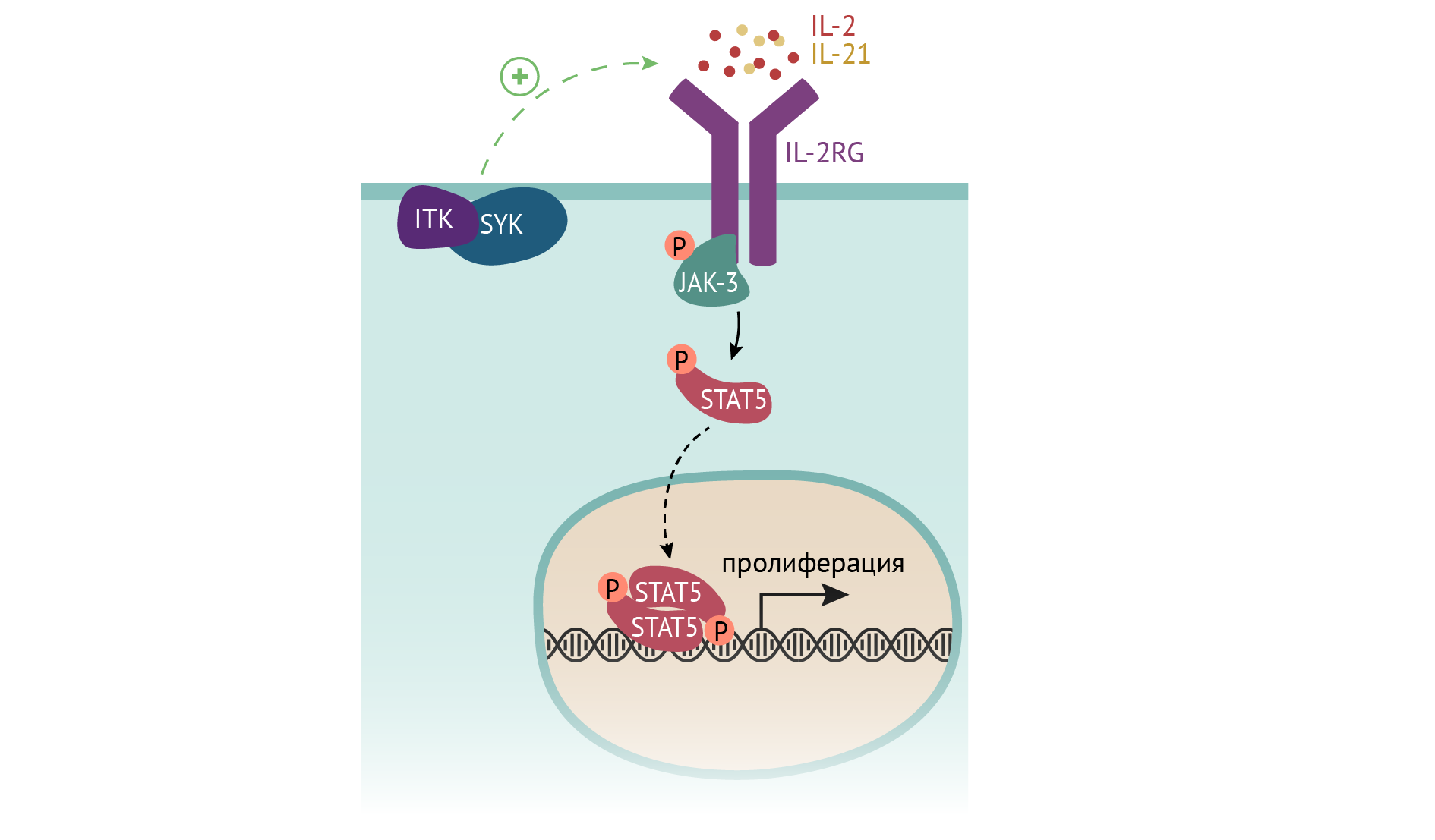
Отдельной группой причин служит дефект рекомбиназ, осуществляющих реаранжировку генома лимфоцитов — того самого процесса, который обусловливает разнообразие спектра антител и Т-лимфоцитарных рецепторов к самым разным антигенам и необходимого для созревания В- и Т-лимфоцитов.
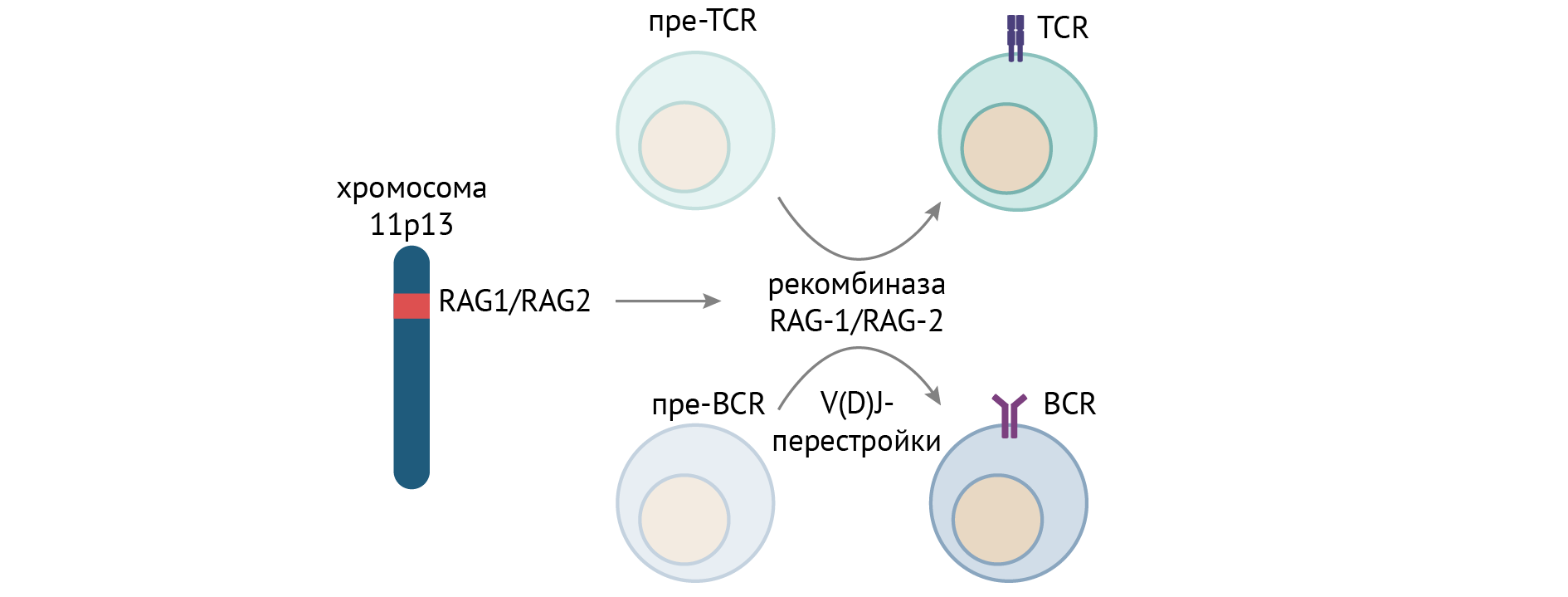
Несмотря на разнообразие причин, приводящих к ТКИД, клинический фенотип один: рецидивирующие и тяжелые инфекции, обусловленные нарушением созревания Т- и В-лимфоцитов [2].
Оротовая ацидурия I/ II типов
Нарушения пиримидинового обмена менее зрелищны. Одним из главных (и в то же время редких) таких нарушений пиримидинового обмена является оротовая ацидурия. Она обусловлена дисфункцией одного или двух ферментов:
- оротатфосфорибозилтрансферазы (превращает оротат в ОМФ);
- оротатфосфорибозилдекарбоксилазы (превращает ОМФ в УМФ) (изолированный дефект данного фермента вызывает II тип оротовой ацидурии, дефект обоих указанных ферментов — оротовую ацидурию I типа) .
Это ферменты конечных этапов синтеза УМФ — предшественника всех остальных пиримидинов. Нетрудно догадаться (если взглянуть на схему ниже и главу 1.2), что данная энзимопатия будет сопровождаться накоплением оротовой кислоты [1, 4, 6].
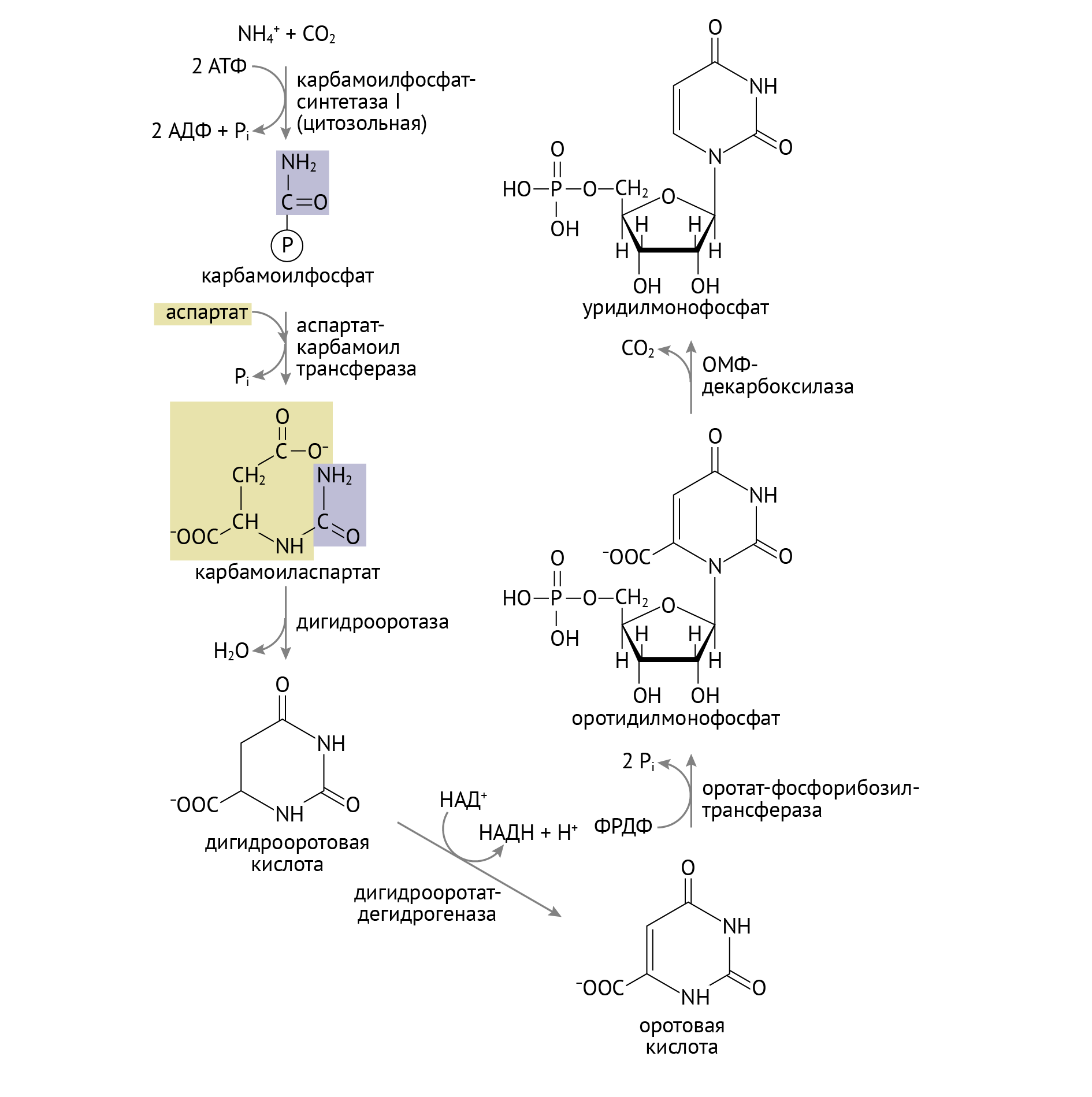
Специфическое клиническое проявление такой энзимопатии — оранжевая моча, которая характерна для первого типа и чье появление обусловлено непосредственным накоплением в ней оротовой кислоты. Но важно не это. А то, что этот дефект чреват нарушением синтеза пиримидинов, а значит, угнетением синтеза нуклеиновых кислот. Характерным будет развитие тяжелой мегалобластной анемии (анемии, обусловленной несовершенным гемопоэзом в результате нарушения репликации ДНК, когда клетка пытается поделиться надвое, а ДНК не хватает). Выглядит это примерно вот так:
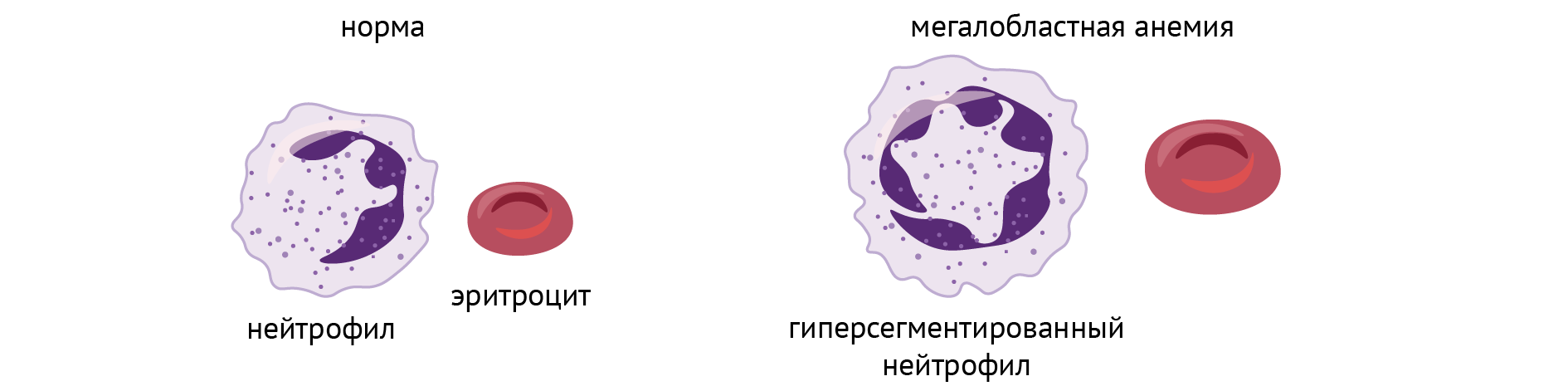
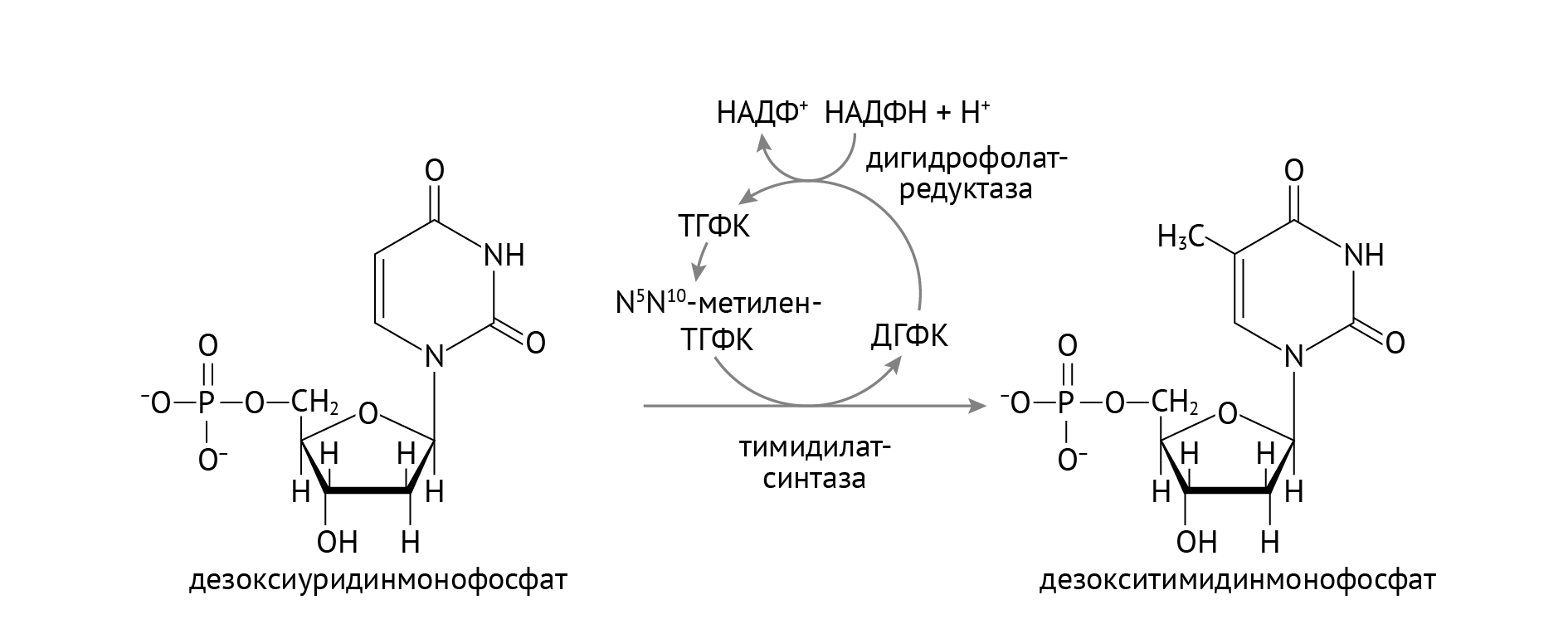
Обычно такое характерно для нехватки витамина В12 и фолиевой кислоты — важных факторов, необходимых для репликации клеток. Их роль заключается в осуществлении превращения дезоксиуридилтрифосфата в дезокситиминтрифосфат — субстрат для ДНК-полимеразы и необходимый компонент для синтеза ДНК. В9 является коферментом тимидилатсинтетазы, а В12 — посредником восстановления активной формы В9 (см. главу 2). Дефицит этих витаминов — частая причина развития мегалобластной анемии. Логично предположить, что лечение такой анемии при оротовой ацидурии будут начинать с их введения, и это справедливо.
Однако эффекта будет чуть меньше, чем никакого. Ибо причина в том, что у нас в принципе нечего превращать в дТТФ. У нас нет уридина в клетках, и он не появится в силу наличия ферментного блока.
Зато разовьется мегалобластная анемия, иммунодефицит, ну и, конечно же, нефролитиаз (кристаллы оротовой кислоты также любят создавать камушки в почечных канальцах).
Выход есть. В таких случаях назначается пожизненное лечение рекомбинантным уридином. По «запасному» пути экзогенный уридин превратится в УМФ и далее по расписанию. Здесь важно вовремя заподозрить возможность наличия у ребенка такого дефекта.
Глава 7 | Сахарный диабет как нарушение всех видов обмена веществ
Источники:
- Кольман Я., Рём К.-Г. Наглядная биохимия., 5-е издание, 2018, с.196.
- Чурилов Л. П., ВасильевА. Г.. Патофизиология иммунной системы, 2014, с. 280–298.
- Mariano Provencio Pulla-Esmo. Handbook of Oncological emergencies, 2016, p. 103–112.
- Цыган В. Н. Патофизиология обмена веществ, учебное пособие. СПБ.: СпецЛит, 2013, с. 53–60.
- Портал «Биохимия для студента». Разделы «Обмен пуринов», «Обмен пиримидинов».
- Тимин О. А. Основы биологической химии, 2018, с. 152–160.
- Елисеев М. С., Барсакова В. Г. Болезнь Лёша—Нихена: клинические проявления и варианты течения, анализ собственного опыта, «Современная ревматология» №3, с. 10.
