
Коронавирусы человека: взаимодействие патоген-хозяин

Тезисы
Коронавирусы человека (HCoV) вызывают респираторные заболевания различной степени тяжести — от легкой до тяжелой. За последние 15 лет мы стали свидетелями появления двух высокопатогенных зоонозных HCoV: коронавирусов ближневосточного респираторного синдрома (MERS-CoV) и тяжелого острого респираторного синдрома (SARS-CoV). Репликация HCoV регулируется множеством факторов организма-хозяина и вызывает резкие изменения в структуре и физиологии клеток. Активация критически важных сигнальных путей во время инфекции HCoV модулирует индукцию противовирусного иммунного ответа и является компонентом патогенеза HCoV. Недавние исследования начали раскрывать некоторые фундаментальные аспекты сложного взаимодействия HCoV с клетками организма человека.
В данном обзоре авторы обобщают современные сведения о факторах вирулентности возбудителей и сигнальных путях, активирующихся вследствие инфицирования HCoV. Отдельно будут рассмотрены вызванные HCoV-инфекцией стрессовая реакция, аутофагия и апоптоз, а также ответ врожденного иммунитета на инфицирование. Также авторы обсуждают перекрестные взаимодействия между различными патологическими путями и изменения «стратегий» вируса в результате реакции организма-хозяина на инфицирование.
Дорогие читатели! Данный текст был опубликован ДО появления SARS-CoV-2, поэтому здесь нет упоминания новой коронавирусной инфекции. Если вам необходимо прочитать непосредственно о вирусологии SARS-CoV-2, то у нас есть переводы двух хороших обзоров, посвященных ему:
Ключевые признаки COVID-19 – https://medach.pro/post/2411
Троица COVID-19: иммунитет, воспаление и вторжение – https://medach.pro/post/2378
Если вы ищите не фундаментальную, а клиническую информацию про COVID-19, то у нас есть переводы двух обновляющихся руководств:
COVID-19: эпидемиология, вирусология, клиническое течение, диагностика и профилактика (UpToDate) – https://medach.pro/post/2366
Рекомендации EMCrit по ведению пациентов с COVID-19 в отделениях интенсивной терапии – https://medach.pro/post/2303
Мы также рекомендуем прослушать три наших подкаста, посвященных COVID-19:
Лабораторная диагностика COVID-19 – https://medach.pro/post/2432
Лучевая диагностика COVID-19 – https://medach.pro/post/2473
Общение с пациентами во время пандемии COVID-19 – https://medach.pro/post/2399
Введение
Коронавирусы — группа оболочечных вирусов с несегментированным геномом, состоящим из единственной плюс-цепи РНК. Помимо заражения множества экономически значимых сельскохозяйственных животных (таких как свиньи и цыплята), известно, что шесть представителей коронавирусов заражают человек, вызывая респираторные заболевания. Среди них коронавирусы тяжелого острого респираторного синдрома (SARS-CoV) и ближневосточного респираторного синдрома (MERS-CoV), которые представляют собой высокопатогенные зоонозные коронавирусы, приведшие в разное время к вспышкам регионального и глобального масштаба.
По данным Международного комитета по таксономии вирусов, коронавирусы относят к порядку Nidovirales, семейству Coronaviridae, подсемейству Coronavirinae. На основании ранних серологических и более поздних геномных исследований представители Coronavirinae делятся на четыре рода: α-, β-, γ- и δ-коронавирусы [126]. Четыре типа вируса (A, B, C и D) относятся к роду β-коронавирусов. Из шести известных человеческих коронавирусов (HCoV) HCoV-229E и HCoV-NL63 относятся к роду α-коронавирусов, тогда как HCoV-OC43 и HCoV-HKU1 относятся к типу A, SARS-CoV — к типу B и MERS-CoV — к типу C рода β-коронавирусов (рис. 1).
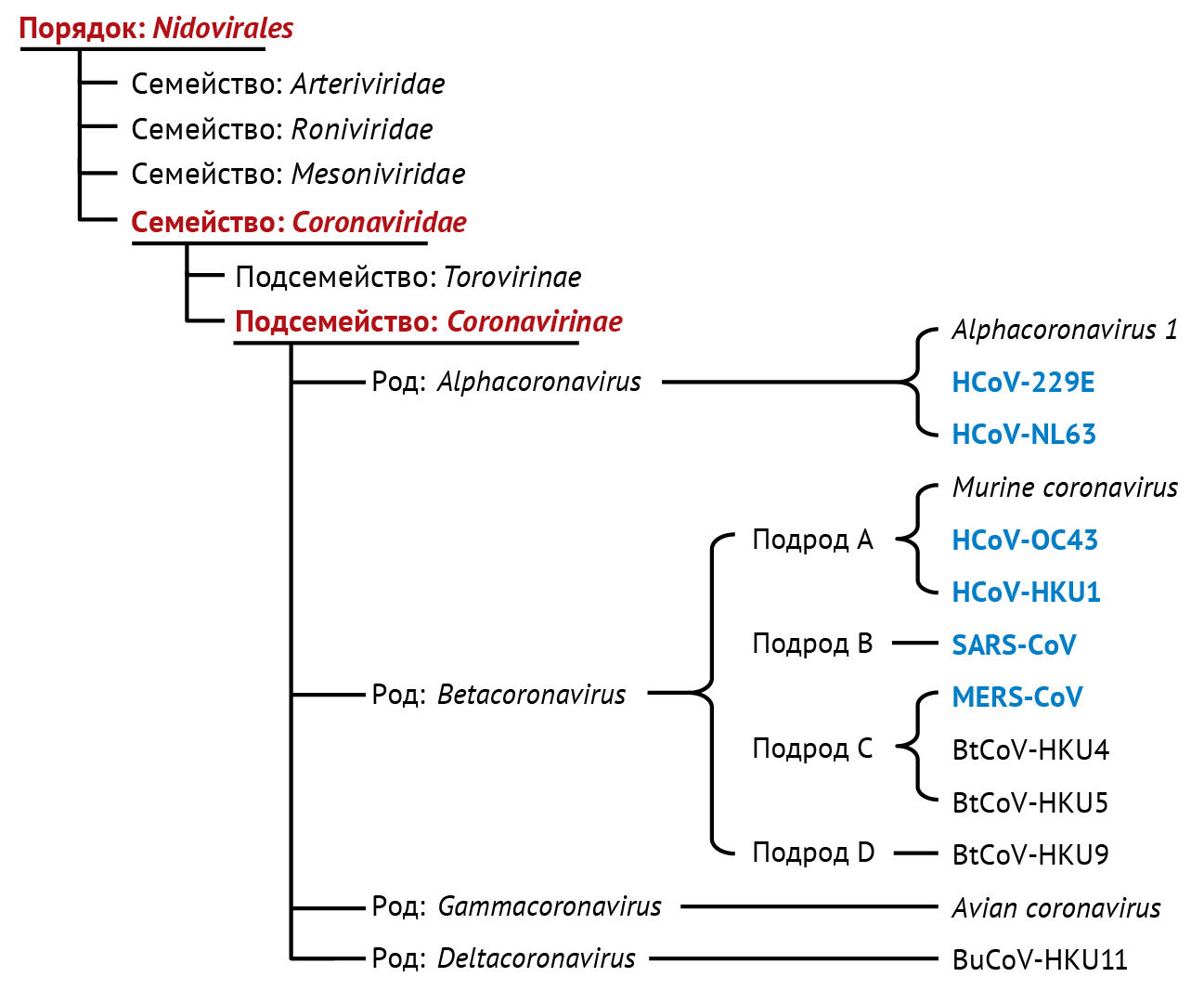
BtCoV — коронавирус летучей мыши;
BuCoV — коронавирус «Bulbul»;
HCoV — человеческий коронавирус;
MERS-CoV — коронавирус ближневосточного респираторного синдрома;
SARS-CoV — коронавирус тяжелого острого респираторного синдрома.
В ноябре 2002 года на юге Китая было впервые выявлено респираторное вирусное заболевание, быстро распространившееся в другие страны и приведшее более чем к 8000 подтвержденных случаев заражения; в конце эпидемии (в июне 2003 года) уровень летальности составлял примерно 9,6 % [98]. Этиологическим агентом был признан SARS-CoV, зоонозный β-коронавирус, который был впервые обнаружен у подковоносых летучих мышей, позже адаптировался для заражения промежуточного хозяина — пальмовой циветы, а затем и человека [64]. После инкубационного периода в 4–6 дней у пациентов развиваются симптомы ОРВИ и пневмонии, которая в тяжелых случаях приводит к летальной дыхательной недостаточности и острому респираторному дистресс-синдрому (ОРДС) [96].
SARS-CoV тропен к тканям многих органов; при его идентификации иммунной системой запускается системный воспалительный ответ, который в случае аберрантности может лежать в основе патогенеза SARS [98]. Несмотря на то, что с 2004 года не было зарегистрировано ни одного случая SARS, богатый генофонд вызывающих его коронавирусов летучих мышей был обнаружен в пещере провинции Юньнань (Китай), что говорит о возможности повторения подобных эпидемий в будущем [50].
В июне 2012 года в Саудовской Аравии появился MERS-CoV — возбудитель респираторного заболевания, подобного SARS [25]. Несмотря на то, что передача вируса между людьми считалась не столь интенсивной, MERS-CoV вызвал две крупные вспышки: в Саудовской Аравии (в 2012 г.) и Южной Корее (в 2015 г.), где общее количество подтвержденных случаев превысило 2000 при уровне летальности 35 % [10]. У пожилых людей, особенно с сопутствующими патологиями, инфекция, вызванная MERS-CoV, имела более тяжелое течение и нередко приводила к летальному исходу [42]. Подобно SARS-CoV, MERS-CoV был обнаружен у летучих мышей, но позже адаптировался к одногорбым верблюдам, которые стали для него промежуточными хозяевами [17]. В настоящее время против SARS-CoV и MERS-CoV не существует одобренных вакцин или специфических противовирусных препаратов.
До появления SARS-CoV были известны только два HCoV: HCoV-229E и HCoV-OC43; оба при введении здоровым добровольцам вызывали слабые симптомы поражения верхних дыхательных путей [45]. Еще два HCoV — HCoV-NL63 и HCoV-HKU1 — были выделены в 2004 и 2005 годах соответственно [31, 127]. Вместе эти четыре всемирно распространенных HCoV, вероятно, ответственны за 15–30 % случаев простудных заболеваний у людей [69]. Несмотря на то, что заболевания, как правило, протекают в достаточно легкой форме, эти штаммы HCoV способны иногда вызывать тяжелые инфекции нижних дыхательных путей у младенцев, пожилых людей или иммунокомпрометированных лиц [41, 97]. Подобно SARS-CoV и MERS-CoV, HCoV-NL63 и HCoV-229E передаются от летучих мышей, тогда как HCoV-OC43 и HCoV-HKU1, вероятно, от грызунов. Важно отметить, что большинство α- и β-коронавирусов, а также коронавирусы, филогенетически связанные с SARS-CoV и MERS-CoV, были обнаружены у различных видов летучих мышей. Следовательно, появляющиеся зоонозные HCoV, такие как SARS-CoV и MERS-CoV, вероятно, берут начало от летучих мышей в результате последовательных мутаций и рекомбинаций. Подвергаясь дальнейшим мутациям при распространении через промежуточных хозяев, они приобрели способность инфицировать людей [22].
В данном обзоре авторы рассматривают цикл репликации HCoV, уделяя особое внимание факторам организма человека, действующим на его отдельных этапах. Затем авторы обобщают современные сведения о важных сигнальных путях, активируемых во время инфекции HCoV, включая реакцию на стресс, аутофагию и апоптоз; характеризуют реакцию врожденного иммунитета на инфицирование. В заключении будут описаны перекрестные взаимодействия между этими путями и изменения в «стратегии», используемой HCoV, под влиянием реакции организма-хозяина.
Репликация HCoV и участие факторов организма человека
Морфология и геномная структура HCoV
Коронавирусы имеют сферическую форму или плеоморфны, их диаметр 80–120 нм. Через электронный микроскоп видно, что поверхность вириона усеяна булавовидными выступами, образованными тримерными шипами (S) из гликопротеина [79]. Более короткие выступы, состоящие из белка димерной гемагглютинин-эстеразы (HE), наблюдаются у некоторых β-коронавирусов (таких как HCoV-OC43 и HCoV-HKU1) [24]. S и HE являются трансмембранными белками типа I с длинным внеклеточным доменом и коротким внутриклеточным доменом. Вирусная оболочка образована мембранным гликопротеином (М) — структурным белком, который встраивается в оболочку и имеет три трансмембранных домена [79]. Кроме того, в нуклеокапсиде в небольшом количестве содержится трансмембранный белок с относительно малой молекулярной массой, известный как белок оболочки (Е) [71]. Наконец, белок нуклеокапсида (N) связывается с геномом РНК в виде «бусинок на нитке», образуя спирально-симметричную структуру [79].
Геном коронавируса представляет собой несегментированный геном, состоящий из единственной плюс-цепи РНК поразительно большого размера (от 27 до 32 тысяч пар оснований). Геномная РНК, имеющая 5’-кэп и поли-(А)-хвост на 3’-конце, содержит множество открытых рамок считывания (ORF). Гены расположены в следующем порядке: 5'-репликаза-S-E-M-N-3' с многочисленными небольшими ORF (кодирующими вспомогательные белки), рассеянными среди структурных генов (рис. 2). Репликаза коронавируса закодирована двумя большими перекрывающимися ORF (ORF1a и ORF1b), которые занимают около двух третей генома и напрямую транслируются из геномной РНК. Тем не менее, структурные и вспомогательные гены транслируются из субгеномных РНК (sg-РНК), которые синтезируются во время транскрипции/репликации генома, как описано ниже.
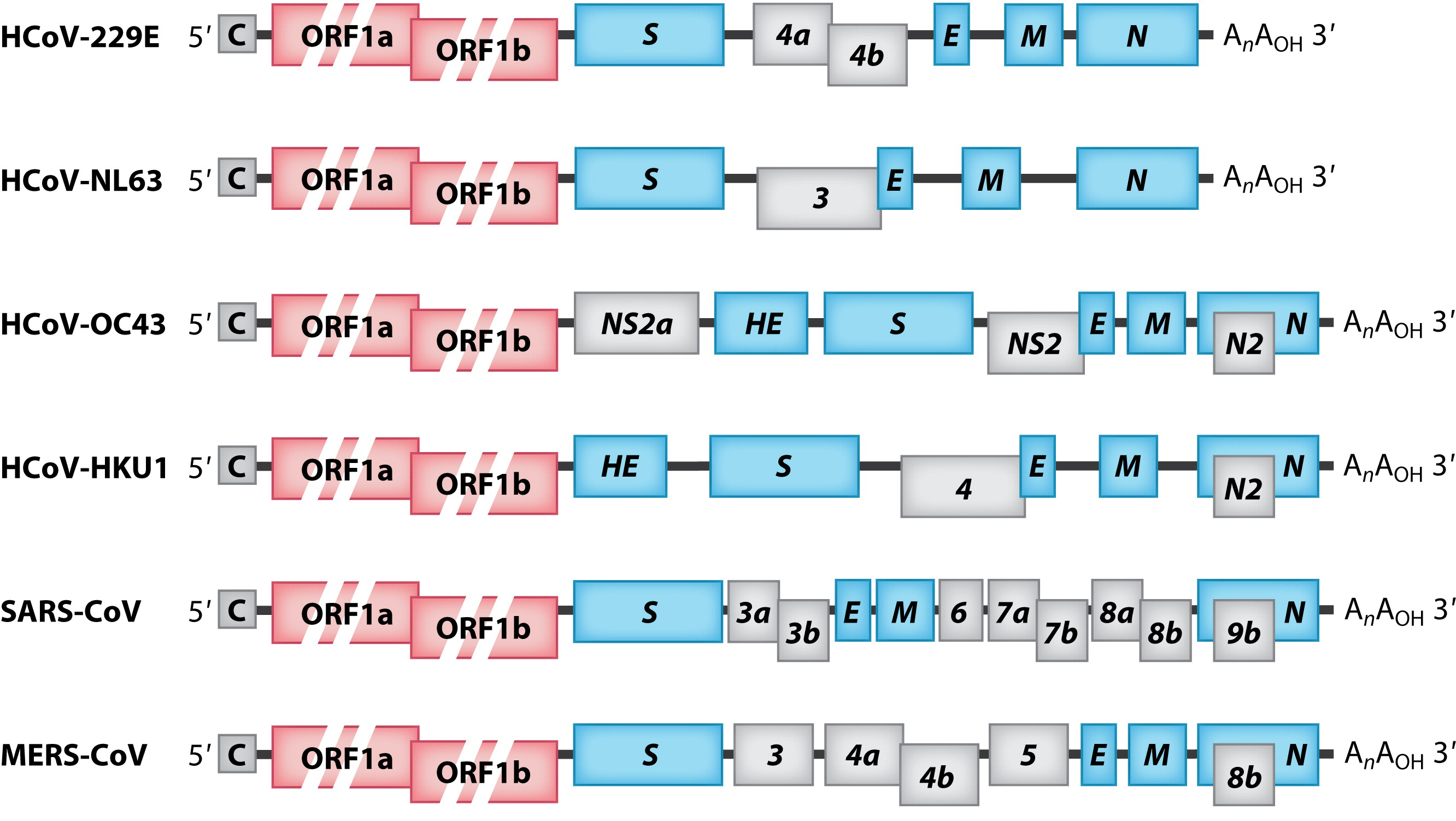
Открытые рамки считывания (ORF1a и ORF1b) представлены в виде коротких красных прямоугольников. Гены, кодирующие структурные шиповидные белки (S), оболочку (E), мембрану (M), нуклеокапсид (N) и гемагглютинин-эстеразу (HE), показаны в виде синих прямоугольников. Гены, кодирующие вспомогательные белки, показаны в виде серых прямоугольников.
Цикл репликации коронавируса разделен на несколько этапов:
- прикрепление и внедрение в клетку;
- трансляция вирусной репликазы;
- транскрипция и репликация генома;
- трансляция структурных белков;
- сборка и высвобождение вириона (рис. 3).
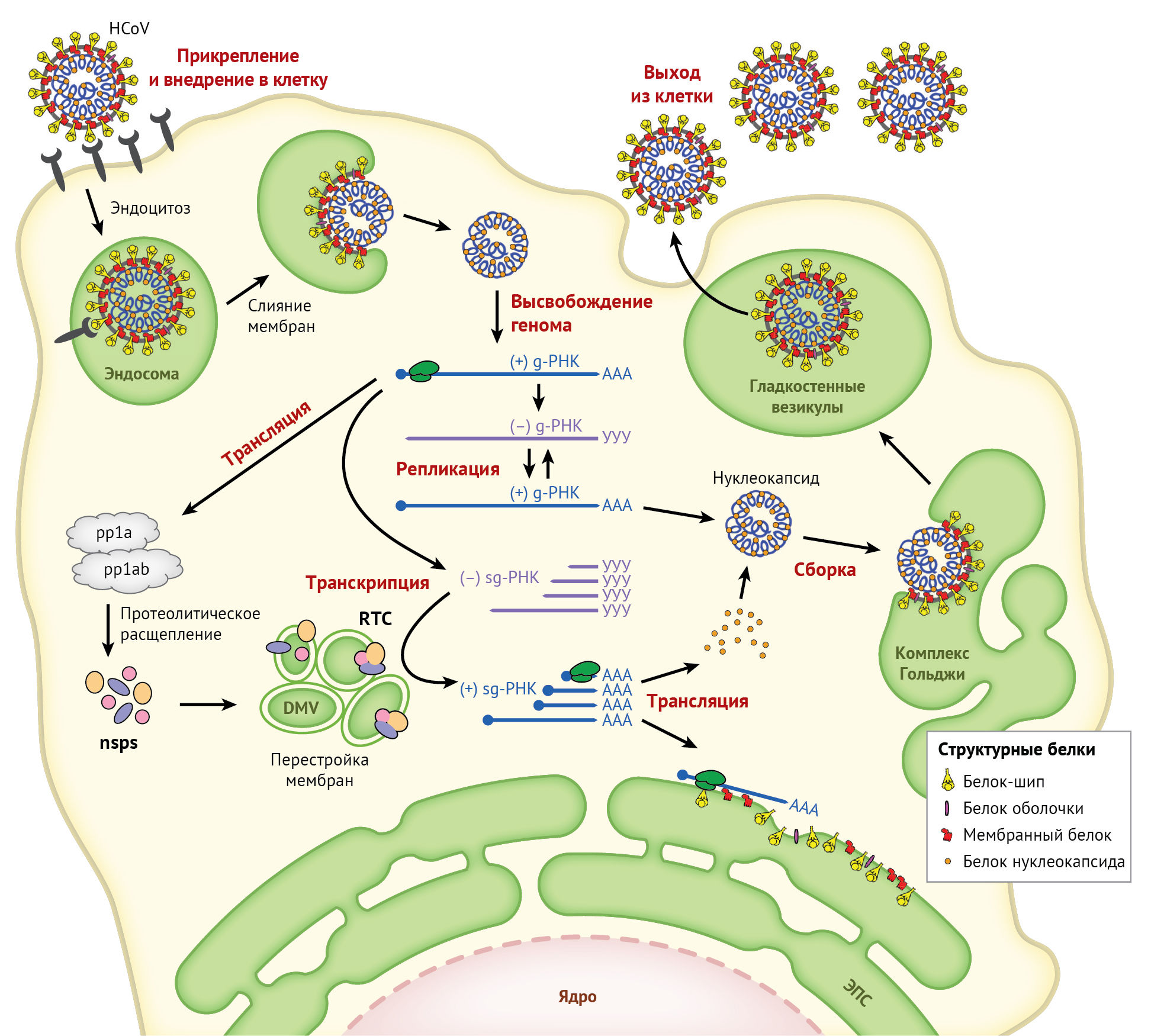
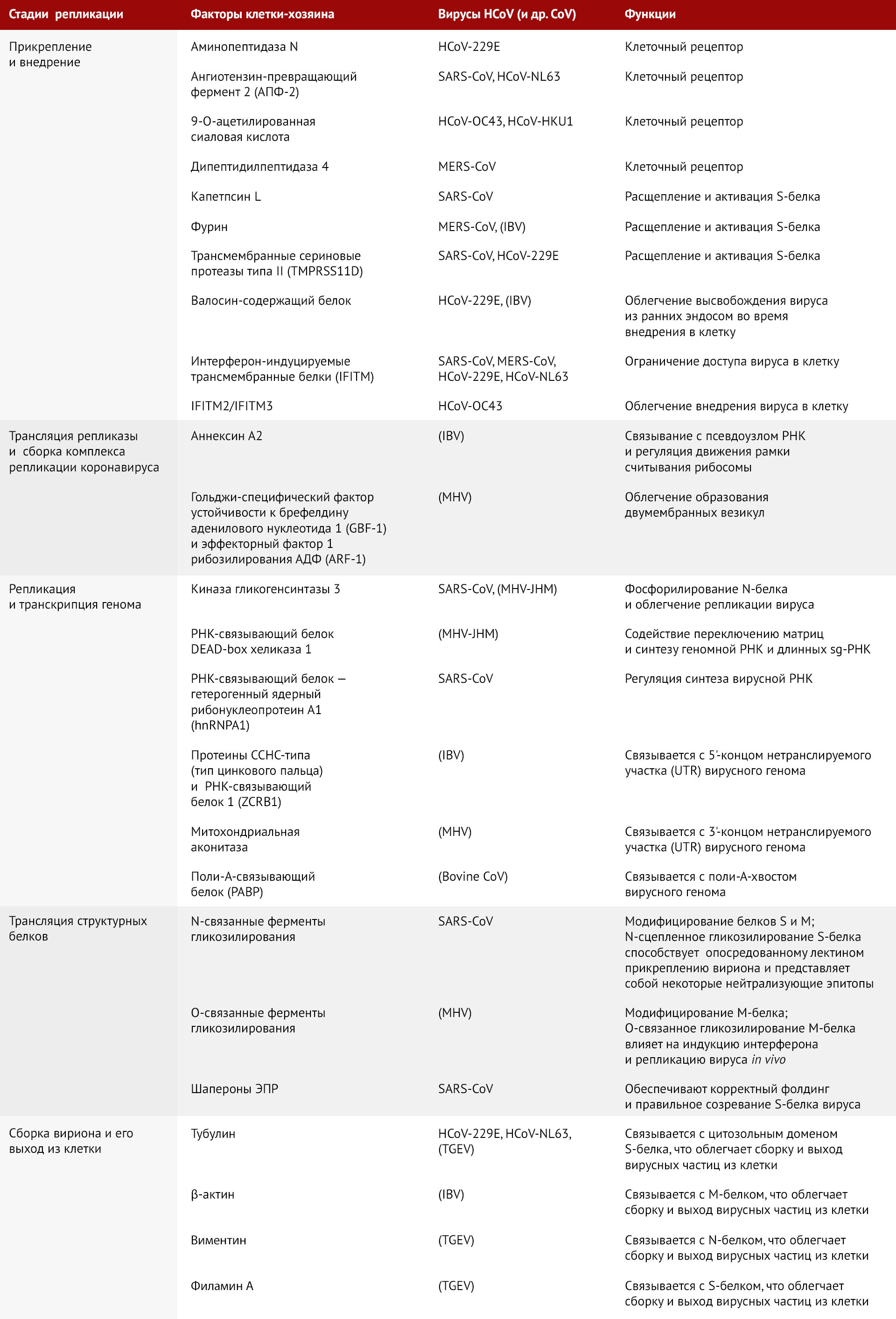
Далее мы кратко рассмотрим каждый этап и обозначим факторы организма-хозяина, вовлеченные в репликацию коронавируса (см. таблица 1).
{kind=link}
Прикрепление и внедрение в клетку
Репликация коронавируса инициируется связыванием S-белка с рецептором(ами) клеточной мембраны. S-белок состоит из двух функциональных субъединиц: S1 («луковица») для связывания с рецептором и S2 («стебель») для слияния с мембраной. Специфическое взаимодействие между S1 и специальным рецептором вызывает резкое конформационное изменение в субъединице S2, приводящее к слиянию вирусной оболочки с клеточной мембраной и проникновению нуклеокапсида в цитоплазму [79]. Взаимодействие с рецептором является определяющим фактором тканевого тропизма коронавируса к клеткам-мишеням человека. Некоторые HCoV используют ферменты клеточной мембраны в качестве рецепторов: HCoV-229E — аминопептидазу N, HCoV-NL63 и SARS-CoV — ангиотензин-превращающий фермент 2 (ACE-2), MERS-CoV — дипептидилпептидазу 4; HCoV-OC43 и HCoV-HKU1 используют в качестве рецептора 9-O-ацетилированную сиаловую кислоту [69].
Расщепление S1 и S2 субъединиц S-белка коронавируса осуществляется одной или несколькими протеазами клетки-хозяина. Например, активация S-белка SARS-CoV требует последовательного расщепления эндосомальной цистеиновой протеазой катепсином L [7, 105] и другой трипсиноподобной сериновой протеазой [4]. С другой стороны, S-белок MERS-CoV содержит два сайта расщепления для фурина, который является повсеместно экспрессируемой протеазой [84]. Интересно, что сайт S1/S2 расщепляется во время синтеза S-белка MERS-CoV, а другой сайт (S2') — во время проникновения вируса. Ранее исследователи открыли аналогичный процесс расщепления у вируса инфекционного бронхита или птичьего коронавируса (IBV) и типичного γ-коронавируса, который инфицирует кур [132]. Кроме того, трансмембранные сериновые протеазы типа II (TMPRSS2 и TMPRSS11D) также участвуют в активации S-белков SARS-CoV [6] и HCoV-229E [5]. Помимо активации S-белка, факторы клетки-хозяина также могут быть вовлечены в последующие стадии проникновения вируса. Например, валозин-содержащий белок способствует высвобождению коронавируса из ранних эндосом, поскольку уменьшение количества этого белка приводит к снижению репликации как HCoV-229E, так и IBV [125].
Факторы клетки-хозяина также могут ограничивать присоединение и проникновение HCoV. Например, интерферон-индуцируемые трансмембранные белки (IFITM) оказывают противовирусное действие широкого спектра в отношении различных РНК-вирусов [2]. Также IFITM ограничивают поступление в клетку SARS-CoV, MERS-CoV, HCoV-229E и HCoV-NL63 [51]. В отличие от них, HCoV-OC43 использует в качестве фактора внедрения IFITM2 или IFITM3 [144]. Недавнее исследование выявило в IFITM несколько аминокислотных остатков, которые контролируют интенсивность внедрения HCoV [145].
Трансляция репликазы и сборка репликационного транскрипционного комплекса
После внедрения и освобождения от капсида геномная РНК служит транскриптом, обеспечивающим кэп-зависимую трансляцию ORF1a для продукции полипротеина pp1a. Кроме того, «скользкая последовательность» и псевдоузел РНК вблизи конца ORF1a позволяют 25–30 % рибосом проскочить один сдвиг рамки, тем самым продолжая трансляцию на ORF1b с образованием более длинного полипротеина pp1ab [79]. При аутопротеолитическом расщеплении pp1a и pp1ab образуется 15–16 неструктурных белков (nsp) (белки, кодируемые вирусом, но не являющиеся частью вирусной частицы — прим. пер.) с различными функциями. Важно отметить, что за активность РНК-зависимой РНК-полимеразы отвечает ген nsp-12 [130], тогда как за активность папаин-подобной протеазы (PLPro) и основной протеазы (Mpro) отвечают гены nsp-3 и nsp-5 соответственно [149]. Гены nsp-3, nsp-4 и nsp-6 также индуцируют перестройку клеточной мембраны с образованием двумембранных везикул (DMV) или сферул [1, 77], где собирается и закрепляется комплекс репликации-транскрипции коронавируса (RTC).
Помимо вторичных структур РНК, запрограммированный рибосомный сдвиг рамки считывания (PRF) может регулироваться вирусными факторами и/или факторами клетки-хозяина. Например, PRF вируса репродуктивно-респираторного синдрома свиней трансактивируется вирусным белком nsp-1β, который взаимодействует с сигналом PRF через предполагаемый РНК-связывающий мотив [65]. Также было доказано, что РНК-связывающий белок хозяина под названием аннексин А2 (ANXA2) формирует структуру псевдоузла в геноме IBV [62].
Считается, что при формировании DMV и сборке RTC факторы клетки-хозяина вовлечены в ранние секреторные пути. Гольджи-специфический, устойчивый к брефелдину-А, гуаниннуклеотид-обменивающий фактор 1 (GBF-1) и его эффекторный фактор 1 рибозилирования AДФ (ARF-1) необходимы как для нормального формирования DMV, так и для эффективной репликации РНК вируса гепатита мыши (MHV) — типичного β-коронавируса, поражающего мышей [119].
Репликация и транскрипция генома
Используя геномную РНК в качестве матрицы, репликаза коронавируса синтезирует полноразмерный антигеном минус-цепи, который, в свою очередь, служит шаблоном для синтеза новой геномной РНК [79]. Также во время прерывистой транскрипции генома полимераза может переключать матрицы в специфических сайтах, которые называются «последовательности, регулируемые транскрипцией», создавая тем самым набор минус-цепей sg-РНК (соответствующих 5’-концу антигенома), которые используются в качестве шаблонов для синтеза набора плюс-цепей sg-РНК (соответствующих 3’-концу генома).
Несмотря на то, что репликация/транскрипция генома в основном опосредуется вирусной репликазой и ограничивается RTC, в этом процессе также принимают участие различные факторы клетки-хозяина. Например, известно, что N-белок коронавируса служит РНК-шапероном и облегчает переключение между матрицами [150, 151]. Важно, что N-белок SARS-CoV и MHV фосфорилируется киназой гликогенсинтазы 3 (GSK-3); было доказано, что ее ингибирование снижает скорость репликации вируса в клетках Vero E6, инфицированных SARS-CoV [129]. Кроме того, GSK-3-опосредованное фосфорилирование N-белка MHV активирует РНК-связывающий белок DEAD-box геликазу 1 (DDX-1), который облегчает считывание матрицы, способствуя синтезу геномной РНК и более длинных sg-РНК [128]. Другой РНК-связывающий белок, под названием «гетерогенный ядерный рибонуклеопротеин А1» (hnRNPA1), также может прочно связываться с N-белком SARS-CoV и регулировать синтез вирусной РНК.
РНК-связывающие белки клетки-хозяина также могут непосредственно связываться с нетранслируемыми областями (UTR) генома коронавируса для изменения скорости репликации/транскрипции. Например, белок ZCRB 1 («цинковый палец CCHC-типа и РНК-связывающий мотив 1») присоединяются к 5'-UTR IBV [111], митохондриальная аконитаза — к 3'-UTR MHV [90], поли-A-связывающий белок (PABP) — к поли-A-хвосту коронавируса крупного рогатого скота [108].
Трансляция структурных белков
Большинство sg-РНК коронавируса являются функционально моноцистронными, и поэтому кэп-зависимым путем транслируется только 5'-ORF [79]. Тем не менее, некоторые sg-РНК для трансляции дополнительных ORF могут использовать и другие механизмы, такие как ослабленное сканирование и внутренняя посадка рибосомы [71]. Трансляция трансмембранных структурных белков (S, HE, M и E) и некоторых связанных с мембраной вспомогательных белков идет в эндоплазматическом ретикулуме, тогда как N-белок транслируется свободными рибосомами в цитозоле [79]. Недавние исследования с использованием рибосомального анализа определили сайты остановки рибосом и выявили несколько коротких ORF, встроенных в сайты, кодирующих вирусный белок [52].
Большинство структурных белков коронавируса подвергаются посттрансляционным модификациям, которые модулируют их функции [40]. Например, белки S и M модифицируются путем гликозилирования [147]. Несмотря на то, что N-связанное гликозилирование S-белка SARS-CoV не способствует связыванию с рецептором [109], оно может участвовать в процессе прикрепления вириона, опосредованном лектином, и составлять некоторые нейтрализующие эпитопы [107]. Кроме того, О-связанное гликозилирование белка М влияет на способность MHV индуцировать репликацию интерферона I типа (IFN-I) у мышей [26]. Правильный фолдинг и созревание вирусных трансмембранных белков (в частности, S-белка) в значительной степени зависят от шаперонов эндоплазматического ретикулума, например, кальнексина [33].
Сборка и высвобождение вириона
Сборка частиц происходит в промежуточных компартментах комплекса Гольджи (ERGIC) и управляется белком М [57, 79]. Гомотипическое взаимодействие белков М обеспечивает основу морфогенеза вирионов, тогда как взаимодействия белков M и S или M и N облегчают привлечение структурных компонентов в место сборки [48]. Е-белок также способствует сборке частиц, взаимодействуя с М-белком и формируя изгибы мембраны [68]. Наконец, частицы коронавируса, поступающие в ERGIC, транспортируются в везикулах и переправляются по секреторному пути для высвобождения путем экзоцитоза.
В сборке и высвобождении вирионов задействованы и другие факторы организма-хозяина. В частности, структурные белки вируса взаимодействуют с цитоскелетом клетки. Взаимодействие тубулинов с цитозольными доменами S-белка коронавирусов HCoV-229E, HCoV-NL63 и TGEV (коронавирус трансмиссивного гастроэнтерита) необходимо для успешной сборки и высвобождения полноценных вирусных частиц [103]. Также было доказано, что взаимодействие М-белка IBV с β-актином, N-белка TGEV с виментином (белком промежуточных филаментов), а также S-белком TGEV с филамином А (актин-связывающим белком) способствует сборке частиц коронавируса и/или их выходу из клетки [121, 143].
Активация аутофагии при инфекции HCoV
Макро-аутофагия (далее — аутофагия) представляет собой консервативный клеточный процесс (внутренние компоненты клетки доставляются внутрь ее лизосом и подвергаются там деградации — прим. пер.). В частности, клетки в стрессовых условиях (голодание, лишение факторов роста или инфицирование патогенными микроорганизмами) инициируют аутофагию в местах отшнуровки везикул эндоплазматического ретикулума, где часть цитоплазмы и/или органелл захватывается аутофагосомами и разрушается при слиянии с лизосомами [135]. Процесс аутофагии регулируется высококонсервативными генами аутофагии (ATG) (рис.4).
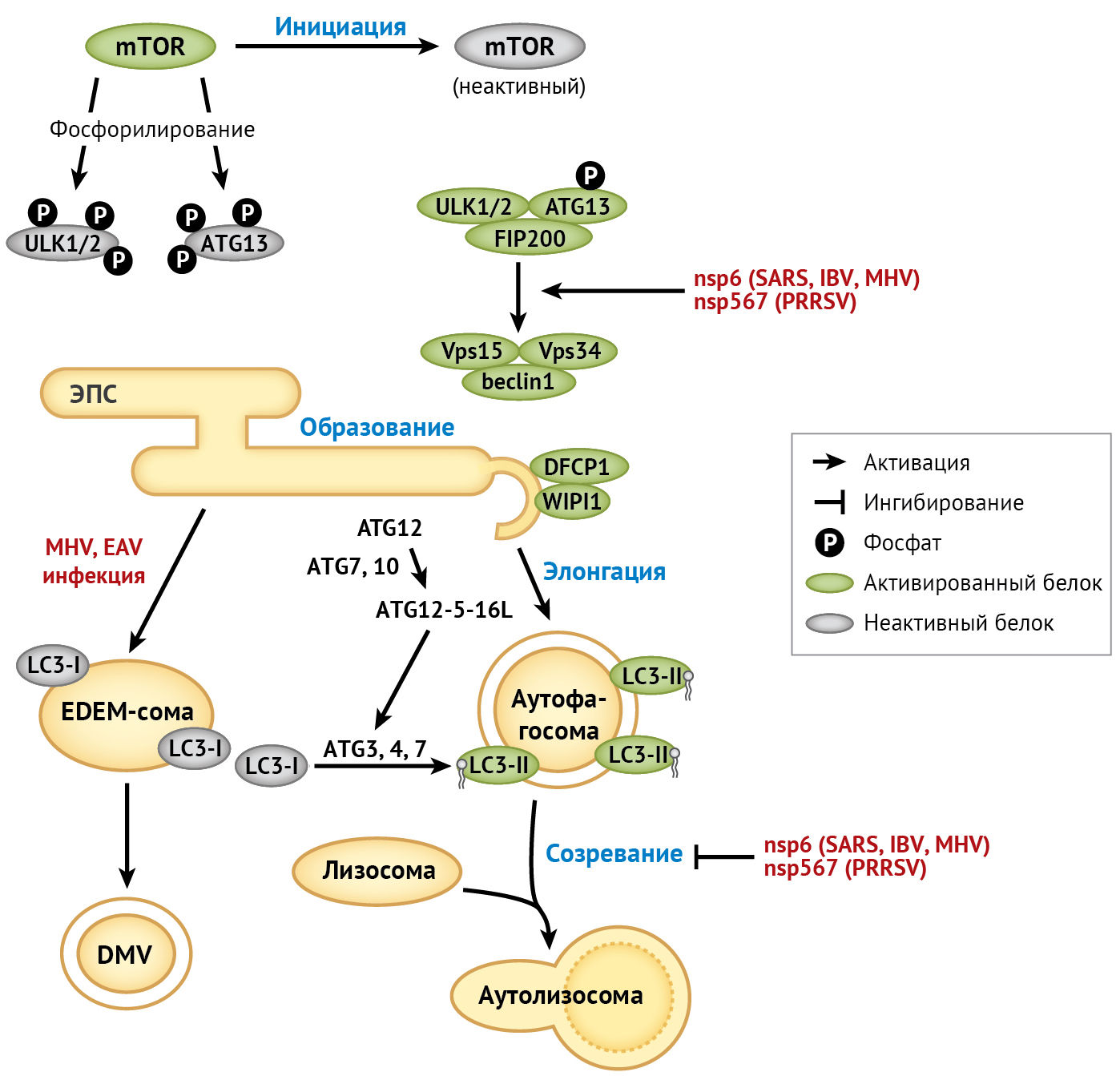
ATG — ген, связанный с аутофагией;
beclin1 — спиральный миозин-подобный белок, взаимодействующий с Bcl-2;
DFCP1 — белок 1, содержащий домен FYVE цинкового пальца; у людей кодируется геном ZFYVE1;
DMV — двумембранная везикула;
EAV — вирус лошадиного артериита;
FIP200 — FAK-семейство (фокальная адгезионная киназа) киназо-взаимодействующих белков массой 200 кДа;
IBV — вирус инфекционного бронхита (птичий коронавирус);
LC-3 — белок 1 легкой цепи 3, связанный с микротрубочками;
MHV — вирус гепатита мыши;
mTOR — белок-мишень рапамицина у млекопитающих;
PRRSV — вирус репродуктивного и респираторного синдрома свиней;
SARS — тяжелый острый респираторный синдром;
ULK — Unc-51-подобная киназа, активирующая аутофагию;
Vps15 — вакуолярная белковая сортировка;
WIPI1 — взаимодействующий с фосфоинозитидом белок 1 с повторяющимся доменом.
Для α-коронавирусной инфекции человека механизм активации аутофагии еще не исследован. У родственного ему α-коронавируса свиней (PEDV) аутофагию активировали в клетках Vero, инфицированных штаммом PEDV CH / YNKM-8/2013, причем ингибирование аутофагии подавляло репликацию вируса и снижало выработку провоспалительных цитокинов [44].
Также активация аутофагии и митофагии (селективное разрушение митохондрий путем аутофагии — прим. пер.) в эпителиальных клетках свиньи (IPEC-J2), инфицированных TGEV (штамм SHXB), способствовала репликации вируса и предотвращала развитие в инфицированных клетках окислительного стресса и апоптоза [148]. Напротив, в двух независимых исследованиях с использованием клеток яичка свиньи, инфицированных TGEV (штамм H165), или клеток IPEC-J2, инфицированных PEDV (штамм SM98), активация аутофагии подавляла репликацию вируса [43, 58]. Такие расхождения в результатах могут возникать из-за использования различных клеточных линий и штаммов вируса, что требует более широкого спектра исследований данной проблемы in vivo.
Что касается β-коронавируса, то в некоторых ранних исследованиях наблюдалась колокализация белков аутофагии LC-3 и Atg-12 с MHV-репликазой nsp-8; этот факт указывает на то, что для формирования DMV могут использоваться компоненты клеточной аутофагии [99]. Однако репликация MHV не оказывала негативного влияния на ATG5−/− эмбриональные фибробласты мыши (MEF). Кроме того, репликация SARS-CoV в MEF была сравнима со сверхэкспрессией ACE-2; это позволяет предположить, что для репликации β-коронавируса не требуется интактная аутофагия [104]. Позже было доказано, что MHV использует механизмы клетки-хозяина для COPII-независимого экспорта везикул эндоплазматического ретикулума при формировании мембран для образования DMV. Этот процесс требует активности не подвергшейся липидизации формы белка LC-3, но не зависит от аутофагии хозяина [101]. Такая независимая от аутофагии активность LC-3 также вовлечена в механизм репликации вируса лошадиного артериита семейства Arteriviridae [89]. Исходя из вышесказанного, вполне вероятно, что другие вирусы порядка Nidovirales также используют LC-3 для репликации.
Коронавирусный белок nsp-6 — это трансмембранный белок, участвующий в образовании DMV при инфицировании SARS-CoV [1]. Сверхэкспрессия nsp-6 в IBV, MHV или SARS-CoV активирует образование аутофагосом в эндоплазматическом ретикулуме через омегасомное промежуточное соединение [18]. Однако аутофагосомы, образование которых было индуцировано инфекцией IBV или сверхэкспрессией коронавирусного белка nsp-6, имеют меньший диаметр по сравнению с аутофагосомами, образование которых вызвано голоданием; этот факт указывает на то, что nsp-6 может ограничивать увеличение диаметра аутофагосом [19].
Индукция апоптоза при инфекции HCoV
Апоптоз является одной из форм запрограммированной гибели клеток, характеризующейся строго контролируемым разрушением клеточных структур, которые встраиваются в мембранные везикулы (известные как апоптотические тела) и поглощаются соседними клетками или фагоцитами [114]. Из-за своей запрограммированной природы апоптоз не является иммуногенным, что отличает его от некротической гибели клеток, при которой неконтролируемая утечка клеточного содержимого активирует воспалительный ответ.
Апоптоз может быть активирован двумя путями (рис. 5). Внутренний путь обусловлен работой белков семейства В-клеточной лимфомы 2 (Bcl-2) [114].
Белки BAX и BAK из этого семейства являются проапоптотическими каналообразующими белками, которые увеличивают проницаемость внешней митохондриальной мембраны (MOMP), тогда как Bcl-2-подобные белки (такие как Bcl-2, Bcl-xL и Mcl-1) являются антиапоптотическими факторами, снижающими проницаемость. В условиях стресса (например, при повреждении ДНК, депривации факторов роста и т. д.) белки домена BH3 активируются для преодоления ингибирующего действия Bcl-2-подобных белков. В результате увеличение МОМР приводит к высвобождению цитохрома С и образованию апоптосомы, а а затем — к активации эффекторной каспазы -3 или -7. Внешний путь апоптоза заключается в связывании лигандов запрограммированной гибели клетки (таких как FasL и фактор некроза опухоли-α (ФНО-α)) с рецепторами гибели на клеточной поверхности (такими как Fas и рецептор 1 ФНО). Это приводит к формированию апоптотического сигнального комплекса и активации каспазы 8, которая вызывает апоптоз либо в результате непосредственной активации эффекторных каспаз, либо посредством перекрестной связи с внутренним путем через активацию белка Bid, содержащего только BH3-домен [114].
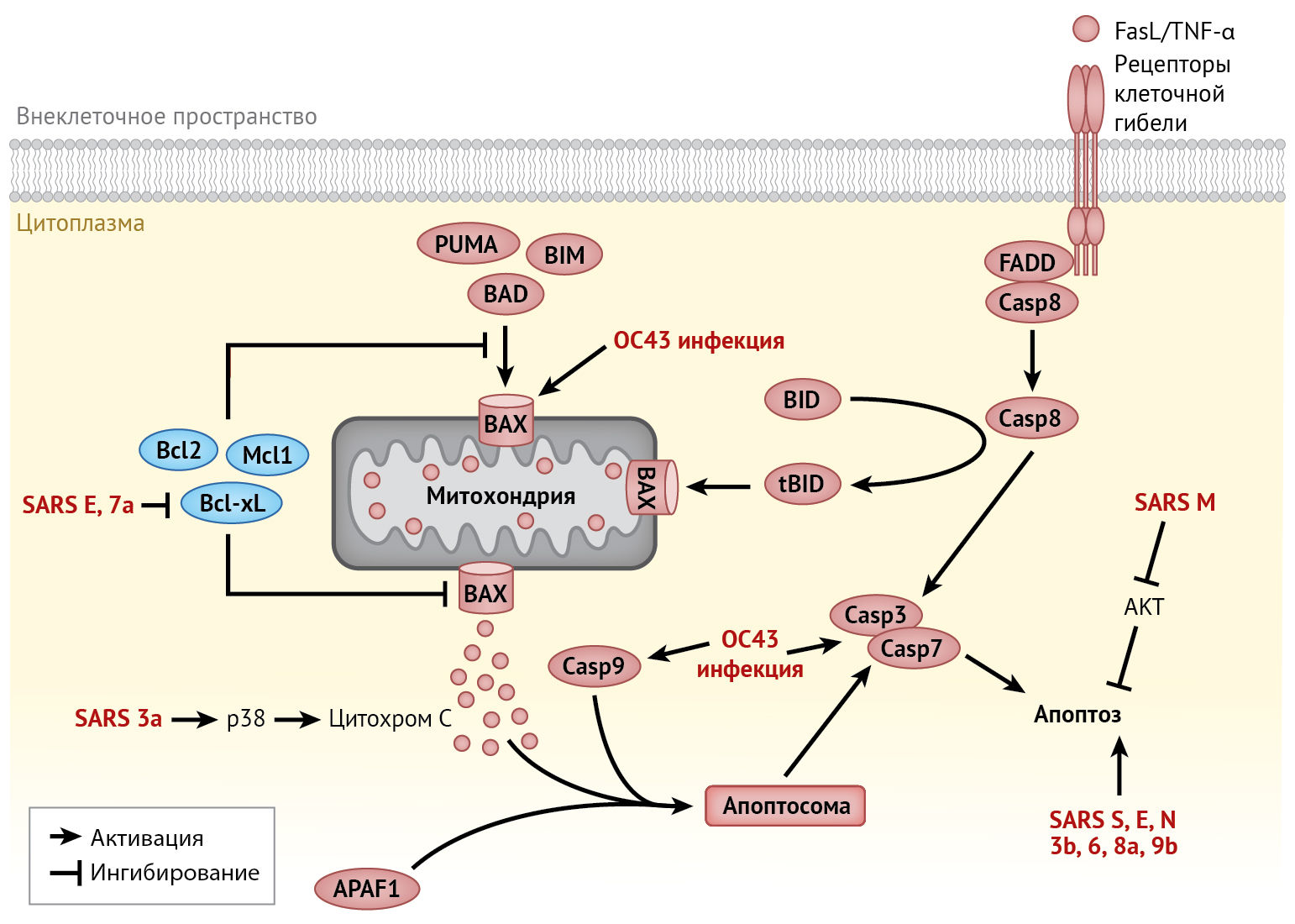
Сокращения:
AKT — RAC-α-серин/треонин-протеинкиназа;
APAF1 — апоптотический пептидаза-активирующий фактор 1;
BAD — Bcl-2-ассоциированный агонист гибели клеток;
BAX — Bcl-2-ассоциированный X-белок;
Bcl-xL — Bcl-2-подобный белок 1;
Bcl2 — В-клеточная лимфома 2;
BID — BH3-взаимодействующий домен агониста клеточной смерти;
BIM — медиатор клеточной смерти, взаимодействующий с Bcl-2;
Casp — каспаза;
FADD — белок взаимодействующий с Fas через домен клеточной смерти;
FasL — Fas-лиганд, трансмембранный белкок типа II, который принадлежит к семейству факторов некроза опухолей;
HCoV — коронавирус человека;
Mcl-1 — миелоидный лейкоз 1;
PUMA — p53-регулируемый модулятор апоптоза;
SARS — тяжелый острый респираторный синдром;
TNF-α — фактор некроза опухолей-α (ФНО-α).
Апоптоз, возникающий при инфицировании HCoV, был тщательно исследован. Признаки апоптоза были обнаружены в тканях легких, селезенки и щитовидной железы, инфицированных SARS-CoV [61]. Кроме того, апоптоз, вызванный инфицированием SARS-CoV, MERS-CoV или другими HCoV, был описан в различных системах in vitro и на животных моделях [113, 136]. HCoV индуцируют апоптоз не только в клетках респираторного эпителия, но и во множестве других типов клеток. Например, HCoV-OC43 индуцирует апоптоз в нейронах [30], тогда как MERS-CoV — в первичных Т-лимфоцитах [15]. Инфицирование HCoV-229E также вызывает массовую гибель дендритных клеток, хотя она и не зависит от индукции апоптоза [82]. Вместе с тем, индукция апоптоза этих иммунных клеток объясняет лимфопению, которая наблюдается при некоторых заболеваниях, вызванных HCoV (например, SARS), а также может способствовать подавлению иммунного ответа хозяина.
В клетках, инфицированных HCoV, апоптоз может быть вызван множеством механизмов. Было доказано, что SARS-CoV индуцирует каспаза-зависимый апоптоз, интенсивность которого обусловлена репликацией вируса. Однако эта зависимость не является существенной, т. к. лечение ингибитором каспаз (z-VAD-FMK) или избыточная экспрессия Bcl-2 не оказывает значительного влияния на репликацию SARS-CoV [36]. Напротив, несмотря на то, что инфекция MERS-CoV первичных Т-лимфоцитов человека имеет абортивное течение, в данном случае апоптоз индуцируется посредством активации как внутреннего, так и внешнего путей [15]. Индукция апоптоза нейронов, инфицированных HCoV-OC43, включает в себя митохондриальную транслокацию BAX, но не зависит от активации каспазы [30].
Апоптоз также индуцируется в клетках, активно экспрессирующих белки SARS-CoV (включая белки S, E, M, N и вспомогательные белки 3a, 3b, 6, 7a, 8a, 9b) [70]. Среди них E-белок и белок 7а SARS-CoV активируют внутренний путь посредством секвестрации антиапоптотического белка Bcl-XL в эндоплазматическом ретикулуме [112]. Другие проапоптотические механизмы в клетках, инфицированных SARS-CoV, включают участие М-белков в передаче сигналов выживания и активацию ионных каналов белками E и 3a [70]. Инфицирование HCoV также модулирует апоптоз путем активации реакции на стресс эндоплазматического ретикулума и митоген-активируемой протеинкиназы (MAPK), что будет подробно рассмотрено ниже.
Активация стресса эндоплазматического ретикулума при HCoV-инфекции
Эндоплазматический ретикулум (ЭР) представляют собой мембранные органеллы для синтеза, фолдинга и модификации секретируемых и трансмембранных белков. Количество белка, синтезируемого в эндоплазматическом ретикулуме, может существенно варьировать под влиянием внеклеточной среды и физиологического состояния организма. Когда способность к фолдингу находится на пределе, белки, еще не прошедшие фолдинг, накапливаются в эндоплазматическом ретикулуме и приводят к стрессу. Во время инфицирования HCoV вирусные структурные белки продуцируются в огромных количествах. В частности, синтез гликопротеина S в значительной степени зависит от белков-шаперонов ЭР и модифицирующих ферментов, отвечающих за его фолдинг и созревание [33]. На самом деле, сверхэкспрессии одного только S-белка SARS-CoV уже достаточно для индукции сильной реакции на стресс эндоплазматического ретикулума [11]. Кроме того, перестройка мембраны для образования DMV и ее истощение при сборке вирионов, также могут способствовать стрессу эндоплазматического ретикулума во время инфицирования HCoV [38].
Для восстановления гомеостаза эндоплазматического ретикулума активируются сигнальные пути, известные как ответ на белки не прошедшие фолдинг (UPR). UPR состоит из трех взаимосвязанных путей, названных в честь трансмембранных белков-рецепторов: протеинкиназы PERK (протеинкиназо-R-подобная ЭР киназа), синтезируемой в эндоплазматическом ретикулуме; инозитол-зависимого фермента 1 (IRE-1) и активирующего фактора транскрипции 6 (ATF-6) (рис. 6). Далее авторы рассматривают механизмы активации данных путей при инфицировании HCoV.
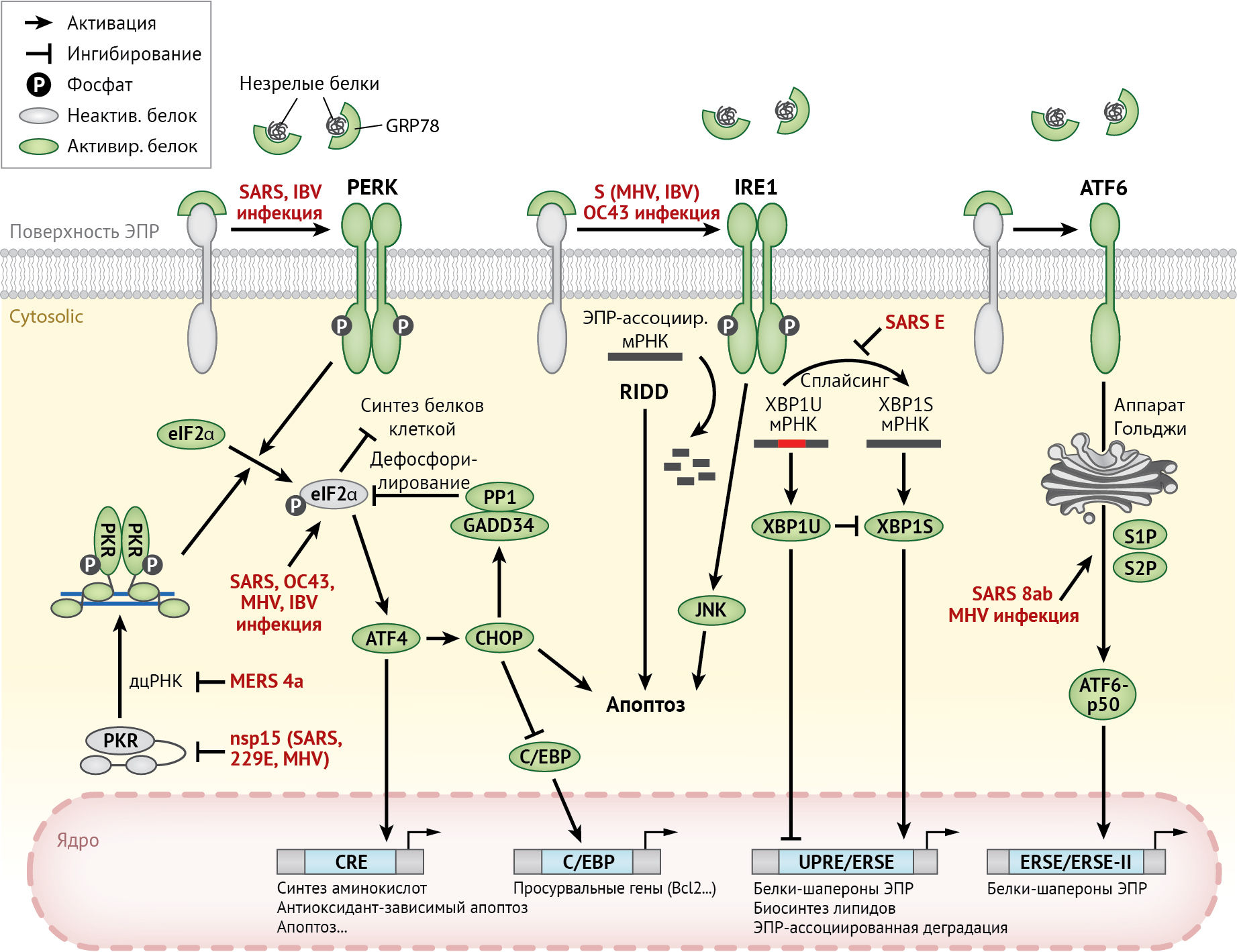
ATF-6 — активирующий фактор транскрипции 6;
C / EBP — CCAAT-энхансер-связывающие белки;
CHOP — C/EBP-гомологичный белок;
CRE — элемент ответа цАМФ;
eIF2α — α-субъединица фактора эукариотической инициации 2;
ERSE — элемент эндоплазматического ретикулума, ответственный за стресс;
GADD-34 — белок 34, индуцируемый повреждением ДНК, который вызывает задержку роста клетки;
GRP-78 — белок массой 78 кДа, регулируемый глюкозой;
HCoV — человеческий коронавирус;
IBV — вирус инфекционного бронхита (птичий коронавирус);
IRE1 — инозитол-зависимый фермент 1;
c-Jun — N-концевая киназа;
MERS — ближневосточный респираторный синдром;
MHV — вирус гепатита мышей;
PERK — протеинкиназа-R-подобная протеинкиназа эндоплазматического ретикулума;
PKR — РНК-активированная протеинкиназа;
PP-1 — протеинфосфатаза 1;
RIDD — IRE-1-зависимый распад мРНК;
SARS — тяжелый острый респираторный синдром;
UPR — развернутый белковый ответ;
UPRE — элемент развернутого белкового ответа;
XBP — X-box-связывающий белок.
PERK-путь и интегрированная реакция на стресс
Среди трех путей UPR первым активируется PERK-путь. При стрессе эндоплазматического ретикулума белок-шаперон GRP-78 связывается с еще не подверженными фолдингу белками и диссоциирует от люминального домена PERK, что приводит к олигомеризации и активации PERK путем аутофосфорилирования. Активированная PERK фосфорилирует α-субъединицу эукариотического фактора инициации 2 (eIF2α), которая ингибирует превращение неактивного ГДФ-связанного eIF2α в активную ГТФ-связанную форму, тем самым подавляя инициацию трансляции. В результате общее снижение синтеза белка уменьшает его приток в эндоплазматический ретикулум и позволяет последнему перестроиться для преимущественной экспрессии генов UPR. Помимо PERK, eIF2α также может фосфорилироваться с помощью трех других киназ: гем-регулируемой ингибиторной киназы (HRI), серин/треониновой-протеинкиназы 2 (GCN-2) и протеинкиназы R (PKR). Последнюю кодирует ген, стимулируемый интерфероном (ISG), который активируется посредством связывания двухцепочечной РНК (дц-РНК) — промежуточного продукта, обычно образующегося при репликации ДНК- и РНК-вирусов.
В целом реакция с участием этих четырех киназ eIF2α и их конвергентных сигнальных путей известна как интегрированная реакция на стресс (ISR) [102].
Несмотря на то, что общий синтез белка при ISR ослабляется, трансляция одной группы генов продолжает поддерживаться. Одним из них является активируемый фактор транскрипции 4 (ATF-4), основной фактор транскрипции «лейциновой молнии» (bZIP), который включает эффекторные гены UPR. ATF-4 также индуцирует другой bZIP-белок — C/EBP-гомологичный белок (CHOP), который отвечает за запуск апоптоза в клетках при длительном стрессе эндоплазматического ретикулума. ATF-4 и CHOP далее вызывают остановку роста и экспрессию белка GADD-34 (индуцируемого ограничением роста и повреждением ДНК) — регуляторной субъединицы протеинфосфатазы 1 (PP-1), которая дефосфорилирует eIF2α. Этот механизм отрицательной обратной связи позволяет возобновить синтез белка после прекращения стресса эндоплазматического ретикулума.
В одном из ранних исследований фосфорилирование PKR, PERK и eIF2α наблюдалось в клетках типа 293/ACE-2, инфицированных SARS-CoV [61]. Удивительно, но снижение активности PKR не повлияло на репликацию SARS-CoV или индуцированное вирусом фосфорилирование eIF2α, несмотря на то, что интенсивность апоптоза, вызванного SARS-CoV, была значительно снижена. Эти данные подтверждают, что активация PKR, вызванная SARS-CoV, может запускать апоптоз независимо от фосфорилирования eIF2α. Как подробно описано в разделе «Врожденный иммунитет и провоспалительный ответ», эндорибонуклеазная активность коронавирусного белка nsp-15 и ds-РНК-связывающая активность белка 4а MERS-CoV также могут подавлять активацию РКR [28, 56, 100].
Активация ISR другими HCoV полностью не изучена. В нейронах, инфицированных HCoV-OC43, на ранней стадии наблюдалось только временное фосфорилирование eIF2α без индукции ATF-4 и CHOP [30].
В отношении коронавирусов, инфицирующих животных, таких как MHV-A59, можно утверждать, что их внедрение в клетку вызывает усиление фосфорилирования eIF2α и активацию ATF-4, но петля отрицательной обратной связи CHOP/GADD-34/PP-1 не активируется, что приводит к продолжительному ослаблению трансляции [3]. Инфицирование TGEV также индуцирует фосфорилирование eIF2α, при этом вспомогательный белок 7 TGEV взаимодействует с PP-1 и смягчает ослабление транскрипции, стимулируя дефосфорилирование eIF2α [21]. Наконец, инфицирование IBV вызывает временное фосфорилирование PKR, PERK и eIF2α на ранних этапах, которое, однако, быстро инактивируется GADD-34/PP-1-опосредованной отрицательной обратной связью [66, 123]. Тем не менее, накопление CHOP способствует IBV-индуцируемому апоптозу, по-видимому, посредством активации проапоптотического белка TRIB-3 и подавления внеклеточной регулируемой киназы 1/2 (ERK-1/2) [66].
Путь IRE-1
IRE-1 (инозитол-зависимый фермент 1 — это трансмембранный белок эндоплазматического ретикулума, который активирует ответ незрелых белков для поддержания функции самого эндоплазматического ретикулума и клетки в целом — прим. пер.) может активироваться посредством диссоциации GRP-78 (как PERK), а также прямым связыванием с N-концевым люминальным доменом белка, еще не подверженного фолдингу [20]. После активации, которая заключается в олигомеризации и аутофосфорилировании, цитозольный рибонуклеазный домен IRE-1 опосредует нетрадиционный сплайсинг мРНК X-box-связывающего белка 1 (XBP-1) [138]. Подвергнувшийся сплайсингу транскрипт кодирует XBP1S — транскрипционный фактор bZIP, индуцирующий экспрессию многочисленных эффекторных генов UPR, которые увеличивают способность эндоплазматического ретикулума к фолдингу [134]. С другой стороны, транскрипт, не подверженный сплайсингу, кодирует XBP1U — крайне нестабильный белок, который снижает активность XBP1S [116]. При длительном стрессе эндоплазматического ретикулума домен IRE-1, обладающий рибонуклеазной активностью, также может разрушать ассоциированные c эндоплазматическим ретикулумом мРНК в процессе, который называется IRE-1-зависимый распад мРНК (RIDD) [49]. Несмотря на то, что RIDD способствует сохранению гомеостаза эндоплазматического ретикулума, уменьшая количество ассоциированной с ним мРНК, деградация мРНК, кодирующей белки выживания, способствует гибели клеток, индуцированной стрессом эндоплазматического ретикулума [81]. Наконец, киназная активность IRE-1 также активирует сигнальный каскад, итогом которого является активация c-Jun N-терминальной киназы (JNK) [118]. Активация пути IRE-1-JNK необходима для индукции аутофагии и апоптоза в клетках при стрессе эндоплазматического ретикулума [93].
В одном из ранних исследований было обнаружено, что сверхэкспрессия S-белка MHV индуцирует сплайсинг мРНК белка XBP-1 [120]. Кроме того, заражение MHV-A59 индуцирует сплайсинг мРНК белка XBP-1, несмотря на то, что белок XBP1S не вырабатывается, по-видимому, из-за подавления трансляции посредством пути PERK/PKR-eIF2α [3]. Напротив, ни инфекция SARS-CoV, ни сверхэкспрессия S-белка SARS-CoV не может индуцировать сплайсинг мРНК белка XBP-1 [27, 120]. Тем не менее, когда ген E-белка SARS-CoV был удален с помощью «обратной генетики», рекомбинантный вирус эффективно индуцировал сплайсинг мРНК XBP-1 и активировал индуцированные стрессом гены, что приводило к более выраженному апоптозу по сравнению с контрольным вирусом дикого типа [27]. Таким образом, E-белок SARS-CoV может служить фактором вирулентности, который подавляет активацию пути IRE-1 и апоптоз, вызванный SARS-CoV. Инфицирование другим β-коронавирусом HCoV-OC43 индуцировало сплайсинг мРНК белка XBP-1 и активацию эффекторных генов UPR [30]. Примечательно, что во время персистирующей инфекции HCoV-OC43 в линиях нервных клеток человека воспроизводимо наблюдались две точечные мутации S-белка. По сравнению с контрольным вирусом дикого типа, рекомбинантный HCoV-OC43, несущий эти две мутации, индуцировал более интенсивные сплайсинг мРНК белка XBP-1 и апоптоз. Таким образом, активация пути IRE-1, по-видимому, способствует запуску апоптоза во время инфицирования HCoV.
Эффективный сплайсинг мРНК белка XBP-1 и активация эффекторных генов UPR также наблюдались в клетках, инфицированных IBV [37]. В отличие от его роли при HCoV-инфекции, в данном случае белок IRE-1 подавлял апоптоз в клетках, инфицированных IBV, предположительно путем преобразования проапоптотического белка XBP1U в антиапоптотический белок XBP1S, а также путем модуляции фосфорилирования ключевых киназ, таких как JNK и AKT (α-серин/треонин-протеинкиназа).
Путь ATF6
Подобно PERK и IRE-1, ATF-6 (активирующий транскрипцию фактор 6 — прим. пер.) активируется диссоциацией белка GRP-78, вызванной стрессом эндоплазматического ретикулума. Кроме того, ATF-6 может активироваться в результате недостаточного гликозилирования или восстановления дисульфидных связей его люминального домена в эндоплазматическом ретикулуме [69]. После активации ATF-6 транслоцируется в аппарат Гольджи, где его N-концевой цитозольный домен (ATF-6-p50) высвобождается в результате расщепления протеазой. Домен ATF-6-p50 представляет собой транскрипционный фактор bZIP, который переносится в ядро и индуцирует экспрессию эффекторных генов UPR, содержащих элемент ответа на стресс эндоплазматического ретикулума (ERSE) или ERSE-II в промоторах [139]. Помимо шаперонов эндоплазматического ретикулума, ATF-6 также индуцирует экспрессию CHOP и XBP-1, тем самым объединяя три пути UPR в единую сигнальную сеть [102].
Активация пути ATF-6 в результате инфицирования HCoV изучена в меньшей степени, и большинство исследований из-за отсутствия специфических антител основывались на косвенных методах, таких как анализ репортерного гена люциферазы. Расщепление белка ATF-6 не обнаружено в клетках, инфицированных SARS-CoV [27], и сверхэкспрессия S-белка SARS-CoV не активирует репортер люциферазы ATF-6 [11]. Однако расщепление ATF-6 и ядерная транслокация наблюдались в клетках, трансфицированных вспомогательным белком 8ab SARS-CoV; также было установлено физическое взаимодействие между белком 8ab и люминальным доменом ATF-6 [110]. Белок 8ab SARS-CoV был обнаружен только в изолятах вируса, полученных на раннем этапе пандемии, в то время как два отдельных белка 8a и 8b были обнаружены в более поздних изолятах и возникли в результате произошедшей в геноме вируса делеции 29 нуклеотидов.
Активация MAPK путей при HCoV инфекции
MAPK (митоген-активируемые протеинкиназы) представляют собой эволюционно консервативные серин/треониновые протеинкиназы, которые активируются в ответ на различные стимулы окружающей среды, такие как тепловой шок, повреждение ДНК, лечение митогенами или провоспалительными цитокинами [55].
MAPK в настоящее время подразделяются на четыре группы: ERK-1/2, ERK-5, p38 и JNK. Для активации MAPK требуется двойное фосфорилирование треонина и тирозина в пределах консервативного мотива TxY с помощью MAPK-киназ (MKK). MКК, в свою очередь, активируются собственными киназами (MKKK, также известными как MAP3K). Активация MAP3K обычно многоэтапная; она регулируется сложными механизмами, такими как аллостерическое ингибирование и/или активация другими киназами (MAP4K). Поскольку MKK обладают высокой субстратной специфичностью по отношению к родственным MAPK, классические сигнальные пути MAPK обычно являются многоуровневыми и линейными. Тем не менее, на некоторых уровнях сигналинга все же имеются перекрестные взаимодействия, и некоторые нетипичные MAPK могут активироваться непосредственно MAP3K. Путем фосфорилирования своих белковых субстратов (во многих случаях являющихся факторами транскрипции) активированные МАРК регулируют многочисленные критические клеточные процессы, такие как пролиферация, дифференцировка, апоптоз и иммунный ответ. Ниже будет рассмотрена активация путей p38, ERK и JNK вследствие инфицирования HCoV (рис. 7).
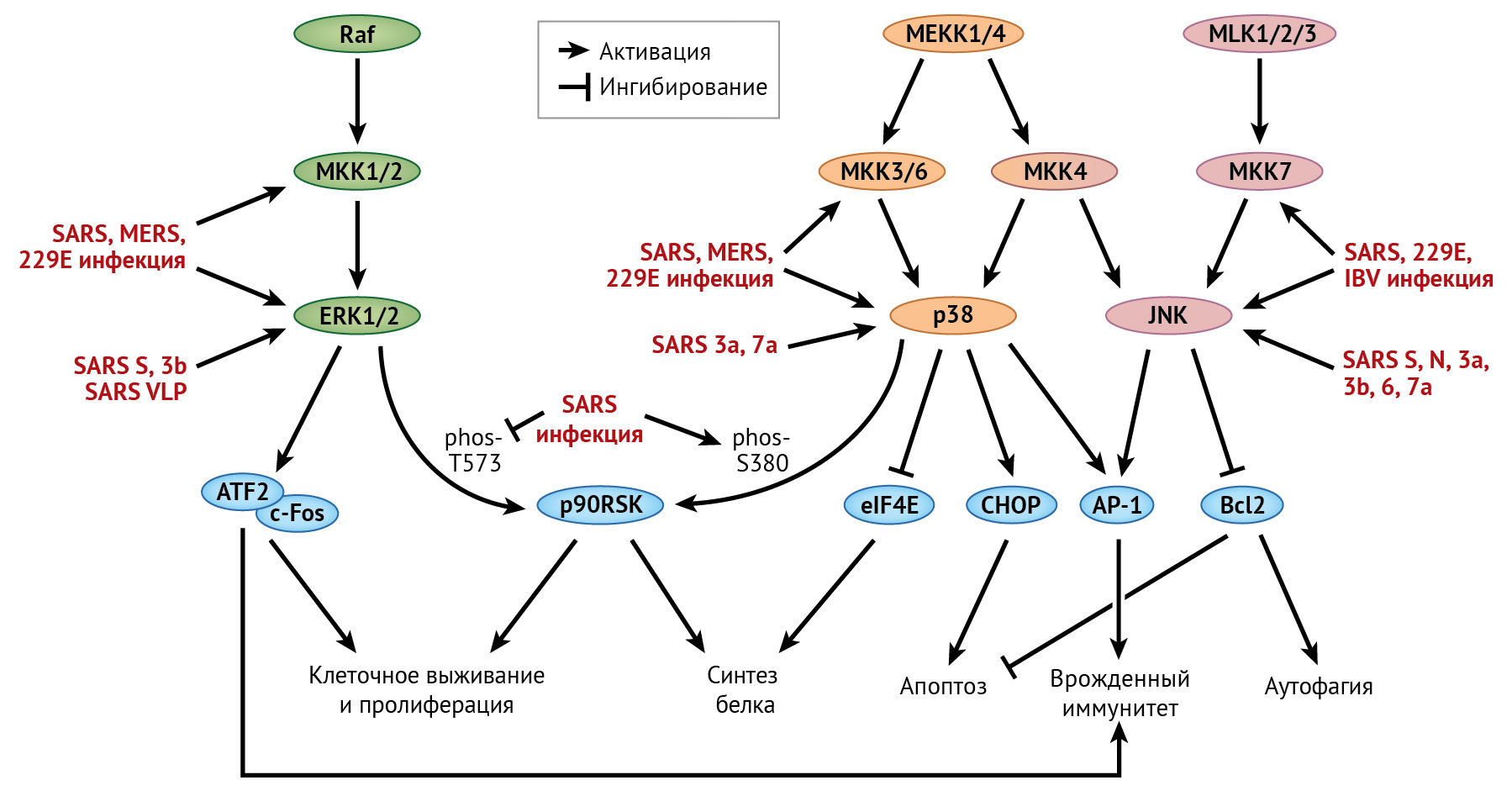
Сокращения:
АР-1 — активирующий пептид 1;
ATF-2 — активирующий фактор транскрипции 2;
Bcl-2 — В-клеточная лимфома 2;
c-Fos — протоонкоген Fos;
CHOP — C/EBP-гомологичный белок;
eIF4E — фактор инициации эукариотической трансляции 4E;
ERK — внеклеточная сигнально-регулируемая киназа;
MAPK — митоген-активируемая протеинкиназа;
MEKK — киназа MAPK/ERK-киназ;
МКК — MAPK-киназа;
MLK — киназа смешанного происхождения;
p90RSK — рибосомальная S6-протеинкиназа 1 с молекулярной массой 90 кДа;
Raf — Raf-1, протоонкоген.
Путь р38
Активированная протеинкиназа р38 транслоцируется в ядро, после чего напрямую или опосредованно фосфорилирует широкий спектр субстратных белков, включая важные факторы транскрипции, такие как белок, связывающийся с цАМФ-зависимым элементом (CREB), ATF-1, преобразователь сигнала и активатор транскрипции 1 (STAT-1) и STAT-3 [140]. Посредством фосфорилирования eIF4E активированный p38 может подавлять инициацию трансляции белка. Путь р38 также может регулировать апоптоз путем фосфорилирования р53 или других проапоптотических белков, таких как ранее упомянутый СНОР [8, 124].
В некоторых исследованиях фосфорилирование р38, его киназы MKK-3/6 и субстратов было обнаружено в клетках Vero E6, инфицированных SARS-CoV [85, 86]. В частности, p38-зависимое фосфорилирование eIF4E может способствовать подавлению синтеза белка в клетке в результате инфицирования SARS-CoV. Однако репликация генома SARS-CoV и синтез вирусного белка не затрагиваются ингибитором p38, что позволяет предположить, что фосфорилирование p38 не является существенным при инфицировании SARS-CoV клеточной культуры [86]. Примечательно, что сверхэкспрессия дополнительного белка 7а SARS-CoV может индуцировать фосфорилирование p38 и ингибировать синтез клеточного белка [60]. Кроме того, активация пути p38 была также вовлечена в апоптоз, вызванный сверхэкспрессией белков 3a или 7a SARS-CoV [60, 95]. Фосфорилирование p38 наблюдалось и в клетках легких плода человека (тип L132), инфицированных HCoV-229E; было обнаружено, что ингибирование p38 подавляет репликацию HCoV-229E [59]. Активация пути p38 также наблюдалась в клетках, инфицированных коронавирусом кошек (FCoV),TGEV, MHV и IBV [34].
ERK путь
ERK — внеклеточная сигнально-регулируемая киназа. Подобно р38, активированная ERK также проявляет свою функцию, фосфорилируя многочисленные транскрипционные факторы, такие как ATF-2, c-Fos и Bcl-6 [137]. В отличие от p38, активированная ERK опосредует фосфорилирование eIF4E-связывающего белка 1 (eIF4EBP-1), вызывая его диссоциацию от eIF4E и тем самым способствуя синтезу белка. ERK также напрямую фосфорилирует рибосомальные протеинкиназы S6 с молекулярной массой 90 кДа (p90RSKs), регулирующие трансляцию белка и пролиферацию клеток [32]. Кроме того, ERK регулирует белки семейства Bcl-2 (например, BAD), тем самым подавляя апоптоз и способствуя выживанию клеток [137].
В одном недавнем исследовании в клетках Vero E6, инфицированных SARS-CoV, наблюдалось фосфорилирование ERK и киназ MKK-1/2 [85]. Фактически, инкубация клеток A549 с S-белком SARS-CoV или вирусоподобными частицами SARS-CoV оказалась достаточной для индукции фосфорилирования киназы ERK [14]. Однако активация p90RSK, одного из ключевых субстратов киназы ERK, затруднена в клетках, инфицированных SARS-CoV [88]. После стимуляции митогеном p90RSK сначала фосфорилируется киназой ERK на С-конце в положении Thr573, что приводит к аутофосфорилированию в положении Ser380. Затем это позволяет связаться с другой киназой, которая фосфорилирует p90RSK на N-конце в положении Ser221, что приводит к ее полной активации [23]. Интересно, что базальный уровень фосфорилирования белка p90RSK в положении Thr573 снижен в SARS-CoV-инфицированных клетках Vero E6 [88]. С другой стороны, фосфорилирование p90RSK в положении Ser380 существенно стимулируется инфекцией SARS-CoV, которая зависит от активации пути p38 [88]. Следовательно, активация p90RSK в SARS-CoV-инфицированных клетках идет по совершенно другому механизму, включающему потенциальные перекрестные контакты между путями киназ ERK и p38. В том же исследовании было выявлено, что лечение ингибитором MKK-1/2 не оказывает влияния на апоптоз, вызванный SARS-CoV; предполагается, что активации пути киназы ERK недостаточно для противодействия апоптозу во время инфицирования SARS-CoV. Этого не происходит в случае IBV, где киназа ERK, по-видимому, служит антиапоптотическим фактором [66]. Наконец, активация пути ERK также наблюдалась в клетках, инфицированных вирусами MERS-CoV и HCoV-229E [69].
Путь JNK
Подобно р38 и ERK, активированная JNK транслоцируется в ядро для фосфорилирования ряда транскрипционных факторов, таких как c-Jun и ATF-2. Затем фосфорилированный c-Jun димеризуется с другими белками с образованием комплекса белка-активатора 1 (AP-1), который связывается с промоторами генов с помощью белка TRE и активирует их экспрессию [47]. Помимо индукции транскрипции проапоптотических генов, таких как Bak и FasL, в ядре JNK также транслоцируется в митохондрии и напрямую фосфорилирует белки семейства Bcl-2, тем самым способствуя запуску стресс-индуцированного апоптоза [133].
Фосфорилирование протеинкиназы JNK и предшествующих ей киназ MKK-4 и MKK-7 наблюдается в клетках Vero E6, инфицированных SARS-CoV [87]. Кроме того, в клетках 293T со сверхэкспрессией S-белка SARS-CoV обнаружено фосфорилирование JNK, опосредованное протеинкиназой C-эпсилон кальций-независимого сигнального пути [72]. Интересен тот факт, что лечение ингибитором JNK устраняет стойкое инфицирование SARS-CoV клеток Vero E6, что свидетельствует о защитной функции пути JNK [87]. Это довольно неожиданно, потому что апоптоз, вызванный сверхэкспрессией N-белка SARS-CoV или вспомогательных белков 6 или 7a, зависит от JNK [69], но в то же время активация JNK способствует IBV-индуцированному апоптозу [37, 39]. Предположительно, JNK может быть проапоптотическим фактором на начальных стадиях инфекции SARS-CoV, но на более поздних этапах переключается на выполнение защитной функции по обеспечению выживания клеток.
Врожденный иммунитет и провоспалительный ответ
Врожденный иммунитет является консервативной стратегией защиты организма, необходимой для первоначального выявления и обезвреживания патогенов, а также последующей активации адаптивного иммунного ответа. Эффективность активации врожденного иммунитета зависит от распознавания патоген-ассоциированных молекулярных паттернов (PAMP) рецепторами распознавания паттернов (PRR), такими как Toll-подобные рецепторы (TLR) и RIG-I-подобные рецепторы (RLR) [69]. После распознавания PAMP рецепторы задействуют адаптерные белки, которые инициируют сложные сигнальные пути с участием множества киназ. Это приводит к активации важных транскрипционных факторов, включая регуляторный фактор интерферона 3 (IRF-3), ядерный фактор κB (NF-κB) и белок-активатор 1 (АР-1). Эти факторы совместно способствуют выработке IFN-I, который высвобождается и действует на соседние клетки, связываясь с рецептором IFN-α/β (IFNAR) [69]. Противовирусная активность IFN-I опосредуется индукцией многочисленных ISG, которые противодействуют репликации вируса различными механизмами (рис. 8). Между тем, индукция цитокинов и хемокинов также активирует воспалительный ответ, который иногда является причиной обширного повреждения тканей и других иммунопатий, возникающих при инфицировании HCoV [98].
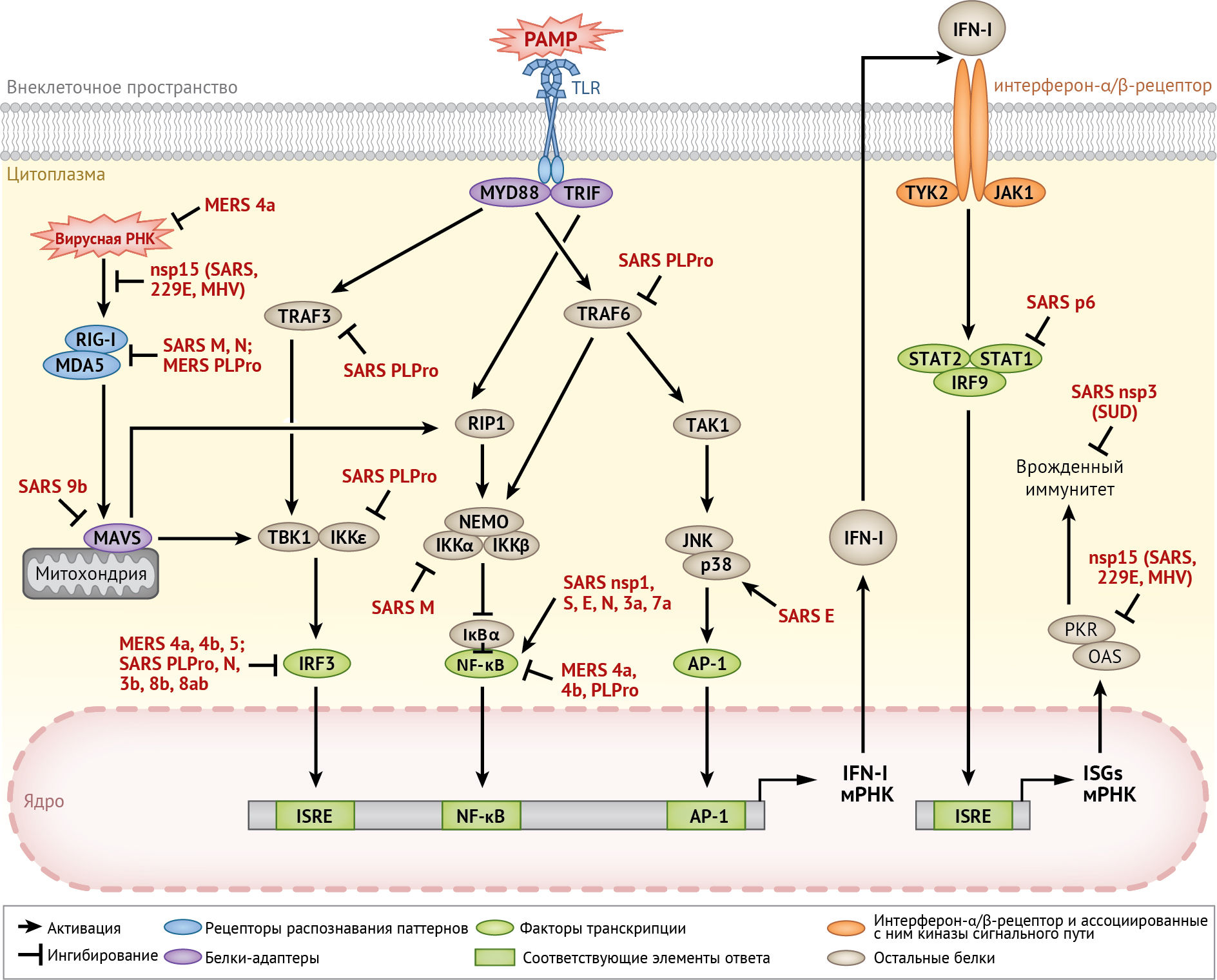
Несмотря на то, что менее опасные HCoV, такие как HCoV-229E, обычно индуцируют высокую степень продукции IFN-I [82], было доказано, что SARS-CoV и MERS-CoV используют многочисленные механизмы для подавления активации врожденного иммунного ответа организма. Некоторые структурные (M и N), неструктурные (nsp-1 и nsp-3) и вспомогательные белки SARS-CoV и/или MERS-CoV являются антагонистами интерферона [40, 69, 70]. Далее будет рассмотрено участие UPR/ISR и MAPK в реакции врожденного иммунитета на инфицирование вирусом гепатита С (HCV), после чего следует разбор двух важных механизмов, используемых HCoV для модуляции врожденного иммунного ответа.
Роль стресса эндоплазматического ретикулума и ISR
Пути UPR могут модулировать передачу сигналов врожденного иммунитета и цитокинов с помощью множества механизмов, включая активацию NF-κB и перекрестное вовлечение путей MAPK [38]. Кроме того, PKR / eIF2α / ATF-4-зависимая активация белка GADD-34 важна для продукции β-интерферона (IFN-β) и интерлейкина 6 (IL-6), индуцированных вирусом Чикунгунья [16]. Кроме того, факторы транскрипции UPR, такие как XBP-1, могут напрямую связываться с промотором/энхансером IFN-β и IL-6 для активации транскрипции [78]. Недавно было обнаружено, что хотя PERK-путь UPR подавлял репликацию TGEV путем активации NF-κB-зависимой продукции INF-I [131], IRE-1-путь способствовал уклонению от действия IFN-I путем снижения уровня экспрессии микроРНК miR-30a-5p [75]. Вопрос о том, будут ли подобные механизмы работать при инфицировании HCoV, требует дальнейшего изучения.
Другим важным антивирусным белком врожденного иммунитета является PKR, для полной активации которой требуется связывание дц-РНК. В недавнем исследовании было обнаружено, что активность эндорибонуклеазы (EndoU), кодируемой коронавирусным геном nsp-15, эффективно подавляет активацию молекул, распознающих дц-РНК клетки-хозяина, включая PKR [56]. Репликация MHV с дефицитом EndoU значительно ослабляется и ограничивается in vivo даже на ранней стадии инфицирования. Это также вызывает повышенный ответ на действие интерферона и обуславливает PKR-зависимый апоптоз [28, 56]. Более того, EndoU-дефицитный коронавирус эффективно активирует MDA-5 и OAS / РНКазу L, вызывает более мягкое заболевание in vivo и стимулирует защитный иммунный ответ [28]. Интересен тот факт, что белок 4а MERS-CoV также был идентифицирован как белок, связывающий дц-РНК [100]. Секвестрируя дц-РНК, белок p4a MERS-CoV подавляет PKR-зависимое ингибирование трансляции, образование стрессовых гранул и активацию передачи сигналов интерферона [100].
Вовлечение MAPK-пути
Пути MAPK вносят вклад в врожденный иммунитет, главным образом, посредством активации АР-1 (активирующего белка 1) и других транскрипционных факторов, регулирующих экспрессию провоспалительных цитокинов. Например, активация p38 необходима для производства цитокинов и развития иммунопатологии у мышей, инфицированных SARS-CoV [53]. Кроме того, активация и высвобождение CCL2 (он же MCP-1 — моноцитарный хемоаттрактантный белок 1 — прим. пер.) и IL-8, вызванные связыванием S-белка SARS-CoV, зависят от активации киназы ERK [12, 14]. Аналогичным образом активация киназы JNK необходима для индукции циклооксигеназы-2 (COX-2) и IL-8 в клетках, сверхэкспрессирующих S-белок SARS-CoV [12, 78]. Подобное участие MAPK-пути в индукции провоспалительных цитокинов (таких как IL-6, IL-8 и TNF-α) было определено и для многих других коронавирусов, инфицирующих животных [34]. Кроме того, MAPK также может регулировать передачу сигналов цитокинов. Например, инфицирование SARS-CoV вызывает дефосфорилирование белка STAT-3 в положении Tyr705 в клетках Vero E6, приводя к его исключению из ядра. Ингибирование киназы p38 частично снижает интенсивность этого процесса, что указывает на ингибирующую роль p38 в передаче сигналов STAT-3 во время инфицирования SARS-CoV [85].
Деубиквитинирующая и ISG-угнетающая активность PLPro HCoV
У коронавирусов одна или две PLPro обычно кодируются в гене белка nsp-3. Помимо полипротеин-расщепляющей протеазной активности, также была выявлена деубиквитинирующая активность для PLPro SARS-CoV, MERS-CoV и IBV, а также для протеолипидного белка 2 вирусов HCoV-NL63 и MHV-A59 [40]. Кроме того, PLPro SARS-CoV и MERS-CoV также распознает белки, модифицированные ISG-15, и катализирует их удаление (ISG-угнетение) [83]. Вероятно, что ISG-угнетение и убиквитинирование критически важных факторов в передаче сигналов врожденного иммунитета используются HCoV для противодействия противовирусному ответу клетки-хозяина. Например, избыточная экспрессия PLPro SARS-CoV или MERS-CoV значительно снижает экспрессию IFN-β и провоспалительных цитокинов в MDA5-стимулированных клетках 293T. Кроме того, PLPro SARS-CoV катализирует убиквитинирование ассоциированного с рецептором TNF фактора 3 (TRAF-3) и TRAF-6, тем самым подавляя действие IFN-I и провоспалительных цитокинов, индуцированных агонистом TLR-7 [63]. Деубиквитинирующая активность PLPro SARS-CoV также подавляет конститутивно активный фосфомиметический фактор IRF-3, что указывает на его участие в постактивации передачи сигналов IRF-3 [80]. Тем не менее, PLPro HCoV может также противодействовать врожденному иммунитету с помощью механизмов, не зависящих от ее деубиквитинирующей и ISG-угнетающей активности [29].
Активность ионных каналов и PDZ-связывающий мотив виропоринов, кодируемых геномом HCoV
Виропорины представляют собой небольшие гидрофобные вирусные белки, которые олигомеризуются с образованием ионных каналов на клеточной мембране и/или вирусной оболочке. Они кодируются геномом широкого спектра вирусов из разных семейств [35]. У коронавирусов была описана активность ионных каналов для Е-белка MHV [76], SARS-CoV [67] и IBV [117]; белков 3a [73] и 8a [13] коронавируса SARS-CoV; белка ORF-3 коронавируса PEDV [122]; белка ORF-4a коронавируса HCoV-229E [141] и белков ns-12.9 коронавируса HCoV-OC43 [142].
Активность ионных каналов необходима для репликации генома некоторых коронавирусов. Например, рекомбинантный IBV, несущий в гене Е мутации T16A или A26F, нарушающие строение ионных каналов, способен к внутриклеточной наработке того же титра вирусных частиц, однако при этом в супернатант высвобождается значительно меньшее количество инфекционных вирионов. Этот факт позволяет предположить, что активность ионного канала может специфически способствовать высвобождению частиц IBV [117]. Точно так же, по сравнению с HCoV-OC43 дикого типа, для рекомбинантного вируса без ns-12.9 наблюдается десятикратное снижение титра in vivo и in vitro [142]. Однако в отличие от IBV, внутриклеточные титры HCoV-OC43-Δns12.9 заметно снижаются; кроме того, электронная микроскопия позволяет предположить наличие дефектов в морфогенезе вириона. Эксперименты с использованием короткой интерферирующей РНК (киРНК) также показали, что снижение концентрации белка 3а SARS-CoV [73], белка ORF-4a HCoV-229E [141] или белка ORF-3 PEDV [122] приводит к ослаблению продукции вирионов и высвобождения соответствующего вируса. Несмотря на то, что активность ионного канала E-белка SARS-CoV не является обязательным условием для репликации вируса, она вносит вклад в его приспособляемость, как показано в сравнительном анализе [91].
Активность ионных каналов также способствует увеличению вирулентности и вносит вклад в патогенез HCoV, в частности, в индукцию стрессового и провоспалительного ответов. В одном раннем исследовании с использованием рекомбинантного вируса, не содержащего гена E, было доказано, что Е-белок SARS-CoV подавляет IRE-1-путь UPR, уменьшает вызванный вирусом апоптоз и стимулирует экспрессию провоспалительных цитокинов [27]. Позже на мутантных формах SARS-CoV с неактивным ионным каналом белка E (EIC-) было доказано, что хотя репликация вируса при этом не затрагивается, вирулентность in vivo на мышиной модели для таких мутантов заметно снижается [91]. Примечательно, что интенсивность отека легких значительно ниже у мышей, инфицированных EIC-отрицательными мутантами, по сравнению с контрольным вирусом дикого типа, что сопровождается снижением продукции провоспалительных цитокинов IL-1β, TNF-α и IL-6 [91]. Активность ионного канала Е-белка SARS-CoV увеличивает проницаемость мембраны комплекса Гольджи и вызывает выход ионов кальция в цитозоль, тем самым активируя NLRP3-зависимое воспаление, стимулирующее выработку IL-1β [92]. Аналогичным образом, у мышей линии BALB/c, интраназально инфицированных HCoV-OC43-Δns12.9, наблюдалось значительное снижение титров вируса и продукции провоспалительных цитокинов IL-1β и IL-6 по сравнению с мышами, инфицированными вирусом дикого типа [142].
Виропорины не только контролируют активность ионных каналов, но и содержат на своем С-конце PDZ-связывающие мотивы (PBM), которые распознаются клеточными PDZ-белками. Например, последние четыре аминокислоты Е-белка SARS-CoV (аспартат-лейцин-лейцин-валин) образуют PDZ-связывающие мотивы, которые взаимодействуют с белком, связанным с протеином PALS1 (протеин, связанный с Lin-7-1), и изменяют его субклеточную локализацию. Это также приводит к изменению структуры плотных контактов и морфогенеза эпителия, что может объяснить нарушение целостности и функции эпителия легких у пациентов с SARS [115]. Важно, что рекомбинантный SARS-CoV с удаленным или мутированным Е-белком PBM ослабляется in vivo и вызывает иммунный ответ пониженной интенсивности по сравнению с контрольным вирусом дикого типа [53]. Обнаружено, что Е-белок PBM SARS-CoV взаимодействует с белком PDZ клетки-хозяина (синтенином) и приводит к его перемещению в цитоплазму, где тот активирует p38 и индуцирует экспрессию провоспалительных цитокинов. Интересно, что при пассаже рекомбинантного SARS-CoV с дефектным Е-белком PBM в клеточной культуре или in vivo, накапливались влияющие на вирулентность обратимые мутации, которые либо восстанавливали Е-белок, либо включали новую последовательность РВМ в гены М или 8a [54]. Это говорит о том, что для вирулентности SARS-CoV необходим по меньшей мере один PBM на трансмембранном белке. Дополнительный белок 3а (другой виропорин, кодируемый геномом SARS-CoV) также содержит C-концевой PBM. Интересно, что несмотря на то, что рекомбинантный SARS-CoV, лишенный как гена E, так и гена 3a, не является жизнеспособным, присутствие любого белка с функциональным PBM может восстановить его жизнеспособность [9]. За исключением Е-белка HCoV-HKU1, все Е-белки HCoV содержат PBM, но их функциональное значение требует более глубокого изучения.
Заключение
Поскольку облигатные внутриклеточные паразиты обладают ограниченными геномными возможностями, все вирусы эволюционировали таким образом, чтобы использовать факторы организма-хозяина для облегчения своей репликации. Между тем, клетки организма-хозяина также создали сложные сигнальные сети для обнаружения, контроля и уничтожения вторгающихся вирусов. Впрочем, эти противовирусные пути часто блокируются различными механизмами вируса. Таким образом, взаимодействие вируса с хозяином представляет собой продолжающуюся эволюционную гонку вооружений на молекулярном и клеточном уровнях. В данном обзоре авторы суммировали недавний прогресс в исследованиях взаимодействия HCoV с клетками организма-хозяина с упором на кооптированные факторы хозяина и критические пути передачи сигналов. Очевидно, что каждый шаг цикла репликации HCoV вовлекает определенные факторы клетки-хозяина; значимые изменения в клеточной структуре и физиологии активируют стрессовый ответ, аутофагию, апоптоз, а также запускают реакцию врожденного иммунитета. С недавним прогрессом в анализе и редактировании генома (таких как CRISPR) очень вероятно, что все больше и больше факторов организма-хозяина и путей, вовлеченных в патогенез коронавирусной инфекции, будут обнаружены и охарактеризованы в недалеком будущем. Использование хорошо изученных моделей животных, инфицированных HCoV, а также систем «обратной генетики» позволят ученым раскрыть ранее неизвестные механизмы, лежащие в основе молекулярной биологии HCoV и их взаимодействия с организмом-хозяином.
С практической точки зрения изучение взаимодействия HCoV с организмом-хозяином также имеет решающее значение в свете потенциального появления в будущем и/или повторного появления высокопатогенного HCoV. За последние 15 лет мы стали свидетелями вспышек двух высокопатогенных зоонозных инфекций HCoV. Тяжелые симптомы, наблюдаемые у пациентов с SARS и MERS, в значительной степени обусловлены иммунопатиями из-за аберрантной активации иммунной системы. Напротив, другие менее опасные формы HCoV вызывают самоограничивающиеся инфекции верхних дыхательных путей, которые довольно редко превращаются в опасные для жизни заболевания у людей с ослабленным иммунитетом. Как эти родственные группы вирусов могут проявлять себя столь различно с точки зрения патогенеза? В определенной степени это можно объяснить различными моделями взаимодействия HCoV с клетками-хозяевами. Одним из примеров является то, что менее опасные HCoV обычно индуцируют высокий уровень продукции IFN-I, тогда как SARS-CoV и MERS-CoV, как известно, противодействуют индукции и передаче сигналов интерферона посредством многочисленных механизмов. Лучшее понимание взаимодействия HCoV — организм-хозяин позволит нам точно определить критические факторы вируса и организма-хозяина, которые контролируют патогенез HCoV, и разработать терапевтические подходы, более эффективные против инфекции HCoV. Например, лекарственные средства, нацеленные на основные факторы хозяина, будут с меньшей вероятностью выбраны для лечения устойчивых к терапии форм HCoV. Кроме того, в то время как сверхактивный иммунный ответ должен быть подавлен при тяжелых коронавирусных инфекциях, усиление активации иммунной системы было бы полезным во время введения вакцины. Наконец, данные о взаимодействии HCoV — организм-хозяин также могут быть экстраполированы на других животных и зоонозные коронавирусы, проливая свет на профилактику и контроль этих значимых в ветеринарном и экономическом отношении патогенов, а также предотвращая появление новых зоонозных коронавирусных инфекций.
